
SEER PEDIATRIC MONOGRAPH
Cancer Incidence and Survival
among Children and Adolescents:
United States SEER Program
1975-1995
This publication was prepared by:
Cancer Statistics Branch
Cancer Surveillance Research Program
Division of Cancer Control and Population Sciences
National Cancer Institute
6130 Executive Blvd.
Executive Plaza North, Room 343J
Bethesda, Maryland 20892-7352
Fax: 301-496-9949
SEER web address: http://www-seer.ims.nci.nih.gov
Suggested citation for the monograph:
Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR (eds).
Cancer Incidence and Survival among Children and Adolescents: United States
SEER Program 1975-1995, National Cancer Institute, SEER Program. NIH
Pub. No. 99-4649. Bethesda, MD, 1999.
Citation for a chapter should also include the chapter authors and chapter title.
This publication and additional data available on the SEER web site:
http://www-seer.ims.nci.nih.gov
Copyright information:
All material in this report is in the public domain and may be reproduced or
copied without permission; citation as to source, however, is appreciated.
Additional Editors
Leslie Bernstein, Ph.D.
Department of Preventive Medicine, University of Southern California/Norris Cancer Center
Charles R. Key, M.D., Ph.D.
New Mexico Tumor Registry
Charles F. Lynch, M.D., Ph.D.
State Health Registry of Iowa
Joseph Simone, M.D.
Utah Cancer Registry
Jennifer Stevens, B.S.
Information Management Services, Inc.
Technical Assistance
Timothy B. Clark, B.A.
Information Management Services, Inc.
Sandra F. Kline
Information Management Services, Inc.
Maureen K. Troublefield
Information Management Services, Inc.
Cancer Incidence and Survival among Children and Adolescents:
United States SEER Program 1975-1995
Editors
Lynn A. Gloeckler Ries, M.S.
Division of Cancer Control and Population Sciences, National Cancer Institute
Malcolm A. Smith, M.D., Ph.D.
Division of Cancer Treatment and Diagnosis, National Cancer Institute
James G. Gurney, Ph.D.
Division of Epidemiology/Clinical Research, Department of Pediatrics,
University of Minnesota
Martha Linet, M.D.
Division of Cancer Epidemiology and Genetics, National Cancer Institute
Thea Tamra, M.D.
Visiting Scientist, Division of Cancer Control and Population Sciences, National
Cancer Institute
John L. Young, Jr., Dr. P.H.
Rollins School of Public Health, Emory University
Greta R. Bunin, Ph.D.
Division of Oncology, University of Pennsylvania School of Medicine and The
Children’s Hospital of Philadelphia

National Cancer Institute
SEER Pediatric Monograph
TABLE OF CONTENTS
i
Page
Foreword ......................................................................................................................... iii
Acknowledgments: Risk Factor Tables ..................................................................... iv
Acknowledgments: SEER ............................................................................................. v
Chapter Contributors................................................................................................... vi
Introduction ................................................................................................................... 1
Lynn A. Gloeckler Ries, Constance L. Percy, Greta R. Bunin
Chapter I Leukemia ..................................................................................................... 17
Malcolm A. Smith, Lynn A.Gloeckler Ries, James G. Gurney,
Julie A. Ross
Chapter II Lymphomas and Reticuloendothelial Neoplasms ............................... 35
Constance L. Percy, Malcolm A. Smith, Martha Linet,
Lynn A. Gloeckler Ries, Debra L. Friedman
Chapter III CNS and Miscellaneous Intracranial and Intraspinal Neoplasms ..................... 51
James G. Gurney, Malcolm A. Smith, Greta R. Bunin
Chapter IV Sympathetic Nervous System Tumors ................................................. 65
Marc T. Goodman, James G. Gurney, Malcolm A. Smith,
Andrew F. Olshan
Chapter V Retinoblastoma .......................................................................................... 73
John L. Young, Jr., Malcolm A. Smith, Steven D. Roffers,
Jonathan M. Liff, Greta R. Bunin
Chapter VI Renal Tumors ............................................................................................ 79
Leslie Bernstein, Martha Linet, Malcolm A. Smith,
Andrew F. Olshan
Chapter VII Hepatic Tumors ...................................................................................... 91
Marc Bulterys, Marc T. Goodman, Malcolm A. Smith,
Jonathan D. Buckley
Chapter VIII Malignant Bone Tumors ...................................................................... 99
James G. Gurney, Andrine R. Swensen, Marc Bulterys
Chapter IX Soft Tissue Sarcomas ............................................................................ 111
James G. Gurney, John L. Young, Jr., Steven D. Roffers,
Malcolm A. Smith, Greta R. Bunin

National Cancer Institute SEER Pediatric Monograph
TABLE OF CONTENTS
ii
Page
Chapter X Germ Cell, Trophoblastic and Other Gonadal Neoplasms .................... 125
Leslie Bernstein, Malcolm A. Smith, Lihua Liu, Dennis Deapen,
Debra L. Friedman
Chapter XI Carcinomas and Other Malignant Epithelial Neoplasms ............... 139
Leslie Bernstein, James G. Gurney
Chapter XII Cancer Among Infants ......................................................................... 149
James G. Gurney, Malcolm A. Smith, Julie A. Ross
Chapter XIII Cancer Among Adolescents 15-19 Years Old .................................. 157
Malcolm A. Smith, James G. Gurney, Lynn A. Gloeckler Ries
Chapter XIV Childhood Cancer Mortality ............................................................. 165
Lynn A. Gloeckler Ries
Chapter XV Other NCI/ NIH Resources .................................................................. 171
International Classification of Childhood Cancer (ICCC) .................................. 175
Index .............................................................................................................................. 179

FOREWORD
iii
iii
National Cancer Institute SEER Pediatric Monograph
Cancer among children is a substantial public concern. Each year in the United States,
approximately 12,400 children and adolescents younger than 20 years of age are diag-
nosed with cancer. Approximately 2,300 children and adolescents die of cancer each
year, which makes cancer the most common cause of disease-related mortality for chil-
dren 1-19 years of age. This monograph assembles under one cover the most detailed
information available on the incidence of childhood cancer in the United States. These
population-based data will be extremely important in furthering our understanding of
the variations in childhood cancer by histologic type and primary site and the variations
in incidence of these cancers over time. The monograph provides information about
childhood cancer incidence and mortality rates that can enhance the level of public
discourse, and it can be used in planning research that will help us to better understand
these cancers and their causes.
Unlike adult cancers that are usually tabulated by primary site, the childhood cancers
are more meaningfully grouped by histologic type and primary site based on the recently
developed International Classification of Childhood Cancer (ICCC). The monograph
details incidence for 1975-1995 and survival by ICCC group and by patient demographic
characteristics. For each of the major ICCC groups, information on known risk factors is
also presented.
The monograph emphasizes not only ICCC group but also age as important factors in
childhood cancer incidence. The cancers discussed include those occurring in children
younger than 15 years of age as well as those occurring in adolescents up to age 19 years.
Some cancers such as neuroblastoma and hepatoblastoma have highest rates among
infants and young children, while others such as Hodgkin’s disease, germ cell tumors
(e.g., testicular cancer) and bone cancers have higher rates among adolescents. It is
important that different distributions of cancer types by age be considered when re-
search programs are developed to improve outcomes for children and adolescents with
cancer.
I would like to thank and congratulate the scientists at the National Cancer Institute
(NCI) and at the various universities and institutions across the United States who
collaborated to make this monograph possible including the Epidemiology and Cancer
Control Strategy Group of the NCI-supported Children’s Cancer Group, which provided
the review of risk factors. I would also like to thank all of the individuals who make the
SEER Program a reality: staff members of the SEER population-based registries, Infor-
mation Management Services, Inc., and NCI. It is through their diligence that these data
have been collected, analyzed, and interpreted. The monograph highlights the impor-
tance of the SEER Program as a national resource. I believe that this document will
prove to be a seminal reference work on childhood cancer for scientists, policy makers
and the public. All of us look forward to the extensive use of this information and the
stimulation of scientific thought that it will engender and ultimately, the reduction of
cancer incidence and mortality in children.
Richard D. Klausner, M.D.
Director
National Cancer Institute

Acknowledgments: Risk Factor Tables
iv
National Cancer Institute SEER Pediatric Monograph
The individuals listed below from the Epidemiology and Cancer Control Strategy Group, of
the NCI-supported Children’s Cancer Group, provided the review of risk factors for selected
cancers. Dr. Greta R. Bunin provided editorial oversight of this effort.
Jonathan D. Buckley, MBBS, Ph.D.
Greta R. Bunin, Ph.D.
Debra L. Friedman, M.D.
Seymour Grufferman, M.D.
Andrew Olshan, Ph.D.
Leslie L. Robison, Ph.D.
Julie Ross, Ph.D.

v
National Cancer Institute SEER Pediatric Monograph
The editors wish to thank the Principal Investigators and the staffs of the contract organi-
zations who provided the cancer incidence data for this report. These organizations, funded
through National Cancer Institute (NCI) contracts, include:
Contracting Organization Principal Investigator
Northern California Cancer Center Dr. Dee W. West
Connecticut State Department of Dr. Anthony P. Polednak
Health Mr. Daniel Savino
Emory University Dr. John L. Young. Jr.
Dr. J. William Eley
Dr. Jonathan M. Liff
University of Hawaii Dr. Laurence N. Kolonel
Dr. Marc T. Goodman
The Fred Hutchinson Cancer Dr. David B. Thomas
Research Center Dr. Beth Mueller
University of Iowa Dr. Charles F. Lynch
Dr. Charles E. Platz
Wayne State University Dr. Linda Weiss
Dr. G. Marie Swanson
University of New Mexico Dr. Charles R. Key
University of Southern California Dr. Ronald K. Ross
Dr. Dennis Deapen
Dr. Leslie Bernstein
University of Utah Dr. Joseph Simone
The production of this report would not have been possible without the efforts of the NCI
staff who ensure the quality and completeness of the SEER data: Benjamin Hankey, Limin
Clegg, April Fritz, Carol Johnson, Carol Kosary, Barry Miller, Constance Percy, Barbara
Ravas, Lynn Ries, Gopal Singh, Thea Tamra (visiting scientist) and Elliott Ware of the
Cancer Statistics Branch and Brenda Edwards of the Cancer Surveillance Research
Program.
Computer support services were provided by Information Management Services (IMS), Inc.
Acknowledgments: SEER

National Cancer Institute SEER Pediatric Monograph
vi
Leslie Bernstein, Ph.D.
Department of Preventive Medicine, University of Southern California/Norris Cancer Center
Jonathan D. Buckley, MBBS, Ph.D.
Department of Preventive Medicine, University of Southern California (Los Angeles)
Marc Bulterys, M.D., Ph.D.
University of New Mexico, currently at Centers for Disease Control and Prevention
Greta R. Bunin, Ph.D.
Division of Oncology, University of Pennsylvania School of Medicine and The Children’s Hospital of Philadelphia
Dennis Deapen, Dr. P.H.
Department of Preventive Medicine, University of Southern California/Norris Cancer Center
Debra L. Friedman, M.D.
Division of Hematology/Oncology, Children’s Hospital and Regional Medical Center, Seattle, WA
Marc T. Goodman, Ph.D.
Cancer Research Center of Hawaii
James G. Gurney, Ph.D.
Division of Epidemiology/Clinical Research, Department of Pediatrics, University of Minnesota
Jonathan M. Liff, Ph.D.
Rollins School of Public Health, Emory University
Martha Linet, M.D.
Division of Cancer Epidemiology and Genetics, National Cancer Institute
Lihua Liu, Ph.D.
Department of Preventive Medicine, University of Southern California/Norris Cancer Center
Andrew F. Olshan, Ph.D.
Department of Epidemiology, University of North Carolina
Constance L. Percy, M.S.P.H.
Division of Cancer Control and Population Sciences, National Cancer Institute
Lynn A. Gloeckler Ries, M.S.
Division of Cancer Control and Population Sciences, National Cancer Institute
Steven D. Roffers, PA, CTR
Rollins School of Public Health, Emory University
Julie A. Ross, Ph.D.
Department of Pediatrics and Cancer Center, University of Minnesota
Malcolm A. Smith, M.D., Ph.D.
Division of Cancer Treatment and Diagnosis, National Cancer Institute
Andrine R. Swensen, M.S.
Division of Epidemiology, University of Minnesota
John L. Young, Jr., Dr. P.H.
Rollins School of Public Health, Emory University
Chapter Contributors
ii

INTRODUCTION
1
National Cancer Institute SEER Pediatric Monograph
INTRODUCTION
Nearly 30 percent of the United States
(US) population is younger than 20 years of
age. Although cancer is rare among those
younger than 20 years of age, it is esti-
mated that approximately 12,400 children
younger than 20 years of age were diag-
nosed with cancer in 1998 and 2,500 died of
cancer in 1998 [1]. As a cause of death,
cancer varies in its relative importance
over the age range from newborn to age 19.
Based on data for 1995, in infants younger
than one year of age, there were fewer than
one hundred cancer deaths (representing
only 0.2% of infant deaths), making it a
minor cause of death in comparison to other
events during the perinatal period. For
children between one and nineteen, cancer
ranked fourth as a cause of death behind
unintentional injuries (12,447), homicides
(4,306), and suicides (2,227). The probabil-
ity of developing cancer prior to age 20
varies slightly by sex. A newborn male has
0.32 percent probability of developing
cancer by age 20, (i.e., a 1 in 300 chance).
Similarly a newborn female has a 0.30
percent probability of developing cancer by
age 20, (i.e., a 1 in 333 chance) [2].
Childhood cancer is not one disease
entity, but rather is a spectrum of different
malignancies. Childhood cancers vary by
type of histology, site of disease origin, race,
sex, and age. To explain some of these
variations, this monograph presents de-
tailed cancer incidence and survival data
for 1975-95, based on nearly 30,000 newly
diagnosed cancers arising in children
during this 21-year interval in the United
States (US). Cancer mortality data col-
lected for the entire US are also shown for
the same time period.
Lynn A. Gloeckler Ries, Constance L. Percy, Greta R. Bunin
MATERIALS AND METHODS (for
definitions and additional details, see
the technical appendix at end of chap-
ter):
Sources of data
The population-based data used in this
monograph for incidence and survival are
from the Surveillance, Epidemiology and
End Results (SEER) Program of the Na-
tional Cancer Institute (NCI) [2]. Informa-
tion from five states (Connecticut, Utah,
New Mexico, Iowa, and Hawaii) and five
metropolitan areas (Detroit, Michigan;
Atlanta, Georgia; Seattle-Puget Sound,
Washington; San Francisco-Oakland,
California; and Los Angeles, California)
comprising about 14% of the United States’
population are used in this monograph.
While Los Angeles did not officially become
a SEER area until 1992, the long standing
cancer registry in Los Angeles provided a
special childhood data file for this study
which included population-based cancer
incidence data back to 1975. This mono-
graph includes 29,659 cancers diagnosed
between 1975 and 1995 in persons younger
than 20 years of age who resided in the
SEER areas listed above: 19,845 cases for
those younger than 15 years of age and
9,814 cases for adolescents aged 15-19
years.
The mortality data are for the same
time period but cover all cancer deaths
among children in the total United States.
Data based on underlying cause of death
were provided by the National Center for
Health Statistics (NCHS).

INTRODUCTION
2
National Cancer Institute
SEER Pediatric Monograph
Table 1: Percent distribution of childhood cancers by ICCC category
and age group, all races, both sexes, SEER, 1975-95
Age
<5 5-9 10-14 15-19 <15 <20
All Sites - Number of cases 9,402 5,024 5,419 9,814 19,845 29,659
%%%%%%
All Sites 100.0 100.0 100.0 100.0 100.0 100.0
36.1 33.4 21.8 12.4 31.5 25.2
Ia - Lymphoid Leukemia 29.2 27.2 14.7 6.5 24.7 18.7
Ia - excl. Acute Lymphoid 0.2 0.3 0.2 0.1 0.2 0.2
Acute Lymphoid 29.0 27.0 14.5 6.4 24.5 18.5
Ib - Acute Leukemia 4.6 4.1 5.4 4.1 4.7 4.5
Ib - excl. Acute Myeloid 1.9 0.9 1.6 0.9 1.5 1.3
Acute Myeloid 2.8 3.2 3.8 3.2 3.2 3.2
Ic - Chronic myeloid leukemia 0.6 0.7 0.9 1.2 0.7 0.9
Id - Other specified leukemias 0.2 0.2 0.1 0.1 0.2 0.2
Ie - Unspecified leukemias 1.4 1.2 0.8 0.5 1.2 1.0
3.9 12.9 20.6 25.1 10.7 15.5
IIa - Hodgkins' disease 0.4 4.5 11.4 17.7 4.4 8.8
IIb - Non-Hodgkins' Lymphoma 2.0 5.2 6.1 6.0 4.0 4.6
IIc - Burkitt's lymphoma 0.8 2.4 1.9 0.6 1.5 1.2
IId - Miscellaneous lymphoreticular
neoplasms
0.4 0.2 0.3 0.2 0.3 0.3
IIe - Unspecified lymphomas 0.3 0.7 0.9 0.7 0.6 0.6
16.6 27.7 19.6 9.5 20.2 16.7
IIIa - Ependymoma 2.6 1.3 1.1 0.5 1.9 1.4
IIIb - Astrocytoma 6.7 14.2 11.8 6.0 10.0 8.7
IIIc - Primitive neuroectodermal tumors 4.3 6.3 3.1 1.0 4.5 3.3
IIId - Other gliomas 2.2 5.0 2.9 1.5 3.1 2.6
IIIe - Miscellaneous intracranial and
intraspinal neoplasms
0.2 0.3 0.3 0.3 0.3 0.3
IIIf - Unspecified intracranial and
intraspinal neoplasms
0.5 0.6 0.4 0.2 0.5 0.4
14.3 2.7 1.2 0.5 7.8 5.4
IVa - Neuroblastoma and
ganglioneuroblastoma
14.0 2.6 0.8 0.3 7.5 5.1
IVb - Other sympathetic nervous system
tumors
0.3 0.1 0.3 0.1 0.3 0.2
6.3 0.5 0.1 0.0 3.1 2.1
9.7 5.4 1.1 0.6 6.3 4.4
VIa - Wilms' tumor, rhabdoid and clear cell
sarcoma
9.7 5.2 0.7 0.2 6.1 4.2
VIb - Renal carcinoma 0.1 0.1 0.4 0.4 0.2 0.2
VIc - Unspecified malignant renal tumors 0.0 0.0 0.0 0.0 0.0 0.0
I(total) - Leukemia
II(total) - Lymphomas and
reticuloendothelial neoplasms
III(total) - CNS and miscellaneous
intracranial and intraspinal
neoplasms
IV(total) - Sympathetic nervous system
V(total) - Retinoblastoma
VI(total) - Renal tumours

INTRODUCTION
3
National Cancer Institute SEER Pediatric Monograph
Table 1 (cont’d): Percent distribution of childhood cancers by ICCC category
and age group, all races, both sexes, SEER, 1975-95
Age
<5 5-9 10-14 15-19 <15 <20
All Sites - Number of cases 9,402 5,024 5,419 9,814 19,845 29,659
%%%%%%
2.2 0.4 0.6 0.6 1.3 1.1
VIIa - Hepatoblastoma 2.1 0.2 0.1 0.0 1.0 0.7
VIIb - Hepatic carcinoma 0.1 0.3 0.5 0.5 0.3 0.3
VIIc - Unspecified malignant hepatic
tumors
0.0 0.0 0.0 0.0 0.0 0.0
0.6 4.6 11.3 7.7 4.5 5.6
VIIIa - Osteosarcoma 0.2 2.2 6.6 4.4 2.4 3.1
VIIIb - Chondrosarcoma 0.0 0.1 0.6 0.6 0.2 0.3
VIIIc - Ewing's sarcoma 0.3 2.1 3.7 2.3 1.7 1.9
VIIId - Other specified malignant bone
tumors
0.1 0.1 0.3 0.3 0.2 0.2
VIIIe - Unspecified malignant bone tumors 0.0 0.1 0.1 0.1 0.1 0.1
5.6 7.5 9.1 8.0 7.0 7.4
IXa - Rhabdomyosarcoma and embryonal
sarcoma
3.4 4.2 2.8 1.9 3.4 2.9
IXb - Fibrosarcoma, neurofibrosarcoma and
other fibromatous neoplasms
1.0 1.4 3.1 3.1 1.7 2.1
IXc - Kaposi's sarcoma 0.0 0.1 0.0 0.1 0.0 0.1
IXd - Other specifed soft-tissue sarcomas 0.7 1.2 2.2 2.1 1.3 1.5
IXe - Unspecifed soft-tissue sarcomas 0.4 0.7 1.0 0.9 0.6 0.7
3.3 2.0 5.3 13.9 3.5 7.0
Xa - Intracranial and intraspinal germ-cell
tumors
0.2 0.8 1.3 0.9 0.7 0.7
Xb - Other and unspecified non-gonadal
germ-cell tumors
1.7 0.1 0.5 1.4 1.0 1.1
Xc - Gonadal germ-cell tumors 1.4 1.1 3.0 9.4 1.7 4.2
Xd - Gonadal carcinomas 0.0 0.0 0.4 1.9 0.1 0.7
Xe - Other and unspecified malignant
gonadal tumors
0.0 0.1 0.1 0.3 0.1 0.1
0.9 2.5 8.9 20.9 3.5 9.2
XIa - Adrenocortical carcinoma 0.2 0.1 0.1 0.1 0.1 0.1
XIb - Thyroid carcinoma 0.1 1.0 3.5 7.4 1.2 3.3
XIc - Nasopharyngeal carcinoma 0.0 0.1 0.7 0.8 0.2 0.4
XId - Malignant melanoma 0.4 0.7 2.0 6.8 0.9 2.9
XIe - Skin carcinoma 0.0 0.0 0.1 0.1 0.0 0.0
XIf - Other and unspecified carcinomas 0.2 0.7 2.5 5.7 1.0 2.5
0.5 0.3 0.6 0.8 0.5 0.6
XIIa - Other specified malignant tumors 0.1 0.1 0.1 0.3 0.1 0.1
XIIb - Other unspecified malignant tumors 0.4 0.3 0.5 0.5 0.4 0.4
VIII(total) - Malignant bone tumors
IX(total) - Soft-tissue sarcomas
X(total) - Germ-cell, trophoblastic and
other gonadal tumors
XI(total) - Carcinomas and other
malignant epithelial
neoplasms
XII(total) - Other and unspecified
malignant neoplasms
VII(total) - Hepatic tumors

INTRODUCTION
4
National Cancer Institute
SEER Pediatric Monograph
In order to calculate rates, population
estimates were obtained from the Bureau
of the Census. In 1990 there were
7,179,865 children residing in the SEER
areas younger than 15 years of age and
9,436,324 younger than 20 years of age. In
the 1990 census, there were about 72
million children younger than 20 years of
age in the whole United States. Twenty-
two percent of the US population is younger
than 15 years of age and an additional 7%
are 15-19 years of age. Annual population
estimates include estimates by 5-year age
groups (<5,5-9,10-14,15-19). Enumeration
of the population at risk by single years of
age was available only for the census years
1980 and 1990. The US Bureau of the
Census provides intercensal population
estimates by 5-year age groups, but not by
single years of age. Therefore, the popula-
tion estimates for 1980 were used in rate
calculations for cases diagnosed from 1976-
84 and the 1990 estimates were used for
cases diagnosed from 1986-94. Whenever
rates by single year of age are shown, the
rates are centered around a decennial
census year, namely, 1976-84 and 1986-94
or the two sets of years combined.
Calculation of rates (see technical appendix)
The incidence and mortality rates are
the annual rates per million person years.
For simplicity, these are labeled as rates per
million. Rates representing more than 5-
years of age are age-adjusted to the 1970
US standard million population. Survival
rates are expressed as percents.
Classification of site and histologic type
The SEER program classifies all cases
by cancer site and histologic type using the
International Classification of Diseases for
Oncology, Second Edition (ICD-O-2) [3]. In
contrast to most cancer groupings, which
are usually categorized by the site of the
cancer, the pediatric classification is deter-
mined mostly by histologic type. The SEER
data have been grouped according to the
International Classification of Childhood
Cancers (ICCC) specifications [4] with a
couple of exceptions for brain cancer.
Please refer to Table 1 for the distribution
by ICCC groupings and age group.
Histologic confirmation
In the SEER program most of the
pediatric cancers (95%) are histologically
confirmed. This is important because most
childhood cancer classifications are based
on histologic types: leukemia, lymphoma,
retinoblastoma, neuroblastoma, etc. The
percentage of histologically confirmed cases,
however, does vary by ICCC category
ranging from a low of 90 percent for the
central nervous system (CNS) (ICCC group
III) to a high of 99 percent for leukemia
(ICCC group I).
OVERVIEW OF CHILDHOOD CANCER
PATTERNS
All sites combined
While grouping all cancer sites to-
gether may be helpful to understanding the
overall cancer burden in young Americans,
it masks the contributions of each primary
site/histology. Therefore, most of the em-
phasis of this monograph is on individual
primary site or histologic groupings; a
separate chapter is shown for each of the
ICCC groupings except group XII which
has few cases.
Overall trends
While the incidence rates for some
forms of childhood cancer have increased
since the mid-1970s, death rates have
declined dramatically for most childhood
cancers and survival rates have improved
markedly since the 1970’s. Each year
approximately 150 children out of every
million children younger than 20 years of
age will be diagnosed with cancer. The

INTRODUCTION
5
National Cancer Institute SEER Pediatric Monograph
overall cancer incidence rate increased from
the mid-1970’s, but rates in the past decade
have been fairly stable (Figure 1). During
the last time period, 1990-95, there is an
indication of a leveling off or slight decline
in the overall incidence rates for each of the
5-year age groups (data not shown). The
overall childhood cancer mortality rates
have consistently declined throughout the
1975-95 time period (Figure 1). Note that
the data are plotted at the mid-year point
throughout this monograph.
Sex
For all sites combined, cancer incidence
was generally higher for males than fe-
males during the 21-year period (Figure 2).
Yet again, an all-sites-combined-rate masks
the sites/histologies for which there is a
female predominance. For some sites/
histologies, there are other factors such as
age where there are differences by sex. For
example, males have somewhat higher
rates of Hodgkin’s disease for children
younger than 15 years of age, but females
have higher rates for adolescents, 15-19
years of age.
Age (5-year age groups)
The average age-specific incidence
rates for each of the four calendar periods
of observation show similar and much
higher cancer rates for the youngest
(younger than 5 years of age) and oldest
(15-19 years of age) age groups than the
two intermediary age groups (Figure 3).
Even though those aged 15-19 years and
those younger than 5 years of age have
similar incidence rates, they have different
mixtures of sites and histologies. The
cancer incidence rates for 5 to 9 year olds
are similar to those seen among 10-14 year
olds.
Age and ICCC group
Fifty-seven percent of the cancers
found among children younger than 20
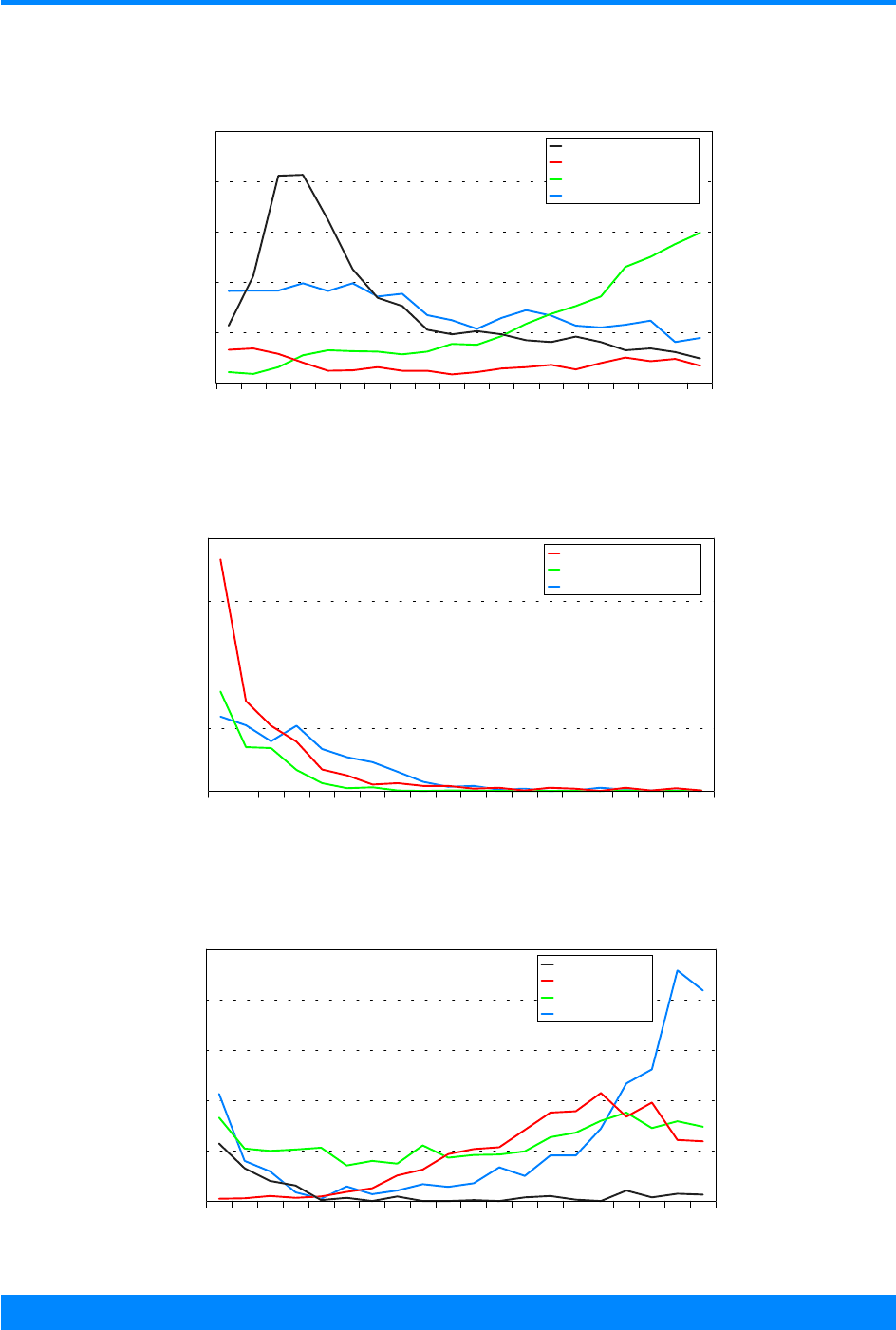
Figure 2: Trends in age-adjusted* incidence rates
for all childhood cancers by sex, age <20
all races combined, SEER, 1975-95
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
))
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
1975 1980 1985 1990 1995
Year of diagnosis
0
50
100
150
200
Average annual rate per million
Male
Female
"
)
*Adjusted to the 1970 US standard population
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
&&
&
&
&
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
1975 1980 1985 1990 1995
Year of diagnosis
0
20
40
60
80
100
120
140
160
180
Average annual rate per million
Incidence
Mortality
,
&
*Adjusted to the 1970 US standard population
Figure 1: Trends in age-adjusted* SEER incidence &
U.S. mortality rates for all childhood cancers
age<20, all races, both sexes, 1975-95

INTRODUCTION
6
National Cancer Institute
SEER Pediatric Monograph
years of age were leukemia, malignant
tumors of the central nervous system (CNS)
or lymphoma. The relative percentage,
however, varied by age group (Table 1).
Leukemia was the most common diagnosis
for those younger than 5, 5-9, and 10-14
years of age but the relative proportion of it
decreased as age increased, from 36 percent
for those younger than 5 years of age to
only 12 percent for adolescents 15-19 years
of age. For 15-19 year olds, lymphomas
were the most common diagnosis, compris-
ing one-fourth of the cases. The second
most common type of cancer was malignant
tumors of the central nervous system for
younger than 5 and 5-9 years of age, and
lymphoma for 10-14 and leukemia for 15-
19 year olds (Table 1).
Figure 4 shows the numbers of cases
used in this study by ICCC group and age.
Leukemia (group I) had the largest number
of cases. Note that these numbers are over
the period 1975 to 1995 for the SEER areas
and do not represent the total number of
childhood cancers in the US in one year.
These numbers indicate the reliability in
the incidence and survival rates, i.e. large
numbers imply stable rates and small
numbers imply unstable rates. Even
though ICCC groups I-III have most of the
cases, there are differences by age group:
group I has more 1-4 year olds, group II has
more 15-19 year olds and group III has
nearly equal numbers for each age group.
There are less than 1,000 cases each in
groups V, VII and XII. Groups VIII-XI tend
to have fewer children younger than 10
years of age compared to 10-19 years of
age.
Incidence by ICCC group
Figure 5 shows the incidence rates per
million children for each of the ICCC
groups. The highest rates are for groups I
(leukemia), II (lymphoma), and III (CNS).
Figure 3: Trends in age-specific incidence rates for
all childhood cancers by age, all races
both sexes, SEER, 1975-95
(
(
(
(
#
#
#
#
"
"
"
"
)
)
)
)
1975-79 1980-84 1985-89 1990-95
Year of diagnosis
0
50
100
150
200
250
Average annual rate per million
<5
5-9
10-14
15-19
)
"
#
(
Figure 4: Number of cases of all childhood cancers
by ICCC and age group, all races
both sexes, SEER, 1975-95
Leukemia - I
Lymphoma - II
Brain/CNS - III
Sympathetic Nerv. - IV
Retinoblastoma - V
Renal - VI
Hepatic - VII
Bone - VIII
Soft tissue - IX
Germ cell - X
Carcinomas - XI
Other - XII
ICCC Group
02468
Number of cases (in thousands)
<1
1-4
5-9
10-14
15-19

INTRODUCTION
7
National Cancer Institute SEER Pediatric Monograph
While the ICCC major groupings indicate
which broad groups of sites/histologies are
important, the sub-groups under each are
necessary to really delineate which histolo-
gies are driving these rates. More detailed
information on the ICCC groups and sub-
groups are contained in other chapters.
Race/ethnicity
For many adult cancers, blacks have
higher incidence rates than whites. For
children, however, black children had lower
incidence rates in 1990-95 than white
children overall and for many of the specific
sites (Figure 6). The time period, 1990-95,
was used for racial/ethnic comparisons
because it was the only time period except
for the decennial census years (1980 and
1990) for which the Census Bureau pro-
vided population estimates for racial groups
other than white and black. The largest
racial difference was for leukemia (ICCC I)
where the rate for whites (41.6 per million)
was much higher than that for blacks (25.8
per million). Cancer incidence rates for
Hispanic children and Asian/Pacific Is-
lander children were intermediate to those
for whites and blacks. The rates for Asian/
Pacific Islanders were similar to whites for
leukemia but lower than whites for CNS
and lymphomas. The incidence rates for
American Indians were much lower than
any other group.
Single year of age
For all sites combined, incidence
varied by age with the highest rates in
infants. The incidence rates declined as age
increased until age 9 and then the inci-
dence rates increased as age increased after
age 9. The pattern, however, varied widely
by ICCC group and single year of age. For
example, high rates were seen among the
very young for retinoblastoma (ICCC group
V) and among adolescents for lymphoma
Figure 5: Age-adjusted* incidence rates for
childhood cancer by ICCC group, age <20, all races
both sexes, SEER, 1975-95
37
24
25
7
3
6
2
9
11
10
14
1
Leukemia - I
Lymphoma - II
Brain/CNS - III
Sympathetic Nerv. - IV
Retinoblastoma - V
Renal - VI
Hepatic - VII
Bone - VIII
Soft tissue - IX
Germ cell - X
Carcinomas - XI
Other - XII
ICCC group
0 1020304050
Average annual rate per million
*Adjusted to the 1970 US standard population
Figure 6: Age-adjusted* incidence rates for
childhood cancer by ICCC group and race/ethnicity
age <20, both sexes, SEER, 1990-95
Am. Indian = American Indian/Native American; API = Asian/Pacific Islander
Hispanic = Hispanic of any race and overlaps other categories
*Adjusted to the 1970 US standard population
41.6
25.8
24.8
41.2
48.5
24.7
18.7
4
14.9
19.5
29.1
25
10.9
19.9
21.8
66.3
55.1
39.9
60.8
55.8
White Black Am. Indian API Hispanic
Race/ethnicity
0
25
50
75
100
125
150
175
Average annual rate per million
I - Leukemia
II - Lymphoma
III - CNS
Other
161.7
124.6
79.6
136.8
145.6
INTRODUCTION
8
National Cancer Institute
SEER Pediatric Monograph
Figure 8
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
(
(
(
(
(
(
(
(
(
(
((
(((((
(
((
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
0 1 2 3 4 5 6 7 8 9 1011121314151617181920
Age (in years) at diagnosis
0
20
40
60
80
Average annual rate per million
Neuroblastoma (IVa)
Retinoblastoma (V)
Wilms' (VIa)
#
(
&
Figure 9
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
&
&
&
&
(
(
(
(
(
(
(
(
(
(
(
((
(
(
(
(
(
(
(
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
#
#
#
#
#
#
#
#
#
##
#
#
#
#
#
#
#
#
#
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
50
Average annual rate per million
Hepatic (VII)
Bone (VIII)
Soft tissue (IX)
Germ cell (X)
#
,
(
&
Age-specific incidence rates for childhood cancer
by ICCC group, all races, both sexes, SEER 1986-94
Figure 7
&
&
&&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
0 1 2 3 4 5 6 7 8 9 1011121314151617181920
Age (in years) at diagnosis
0
20
40
60
80
100
Average annual rate per million
Ac. Lymph. Leuk (Ia)
Ac. Myeloid Leuk (Ib)
Lymphoma (II)
Brain/CNS (III)
#
,
(
&

INTRODUCTION
9
National Cancer Institute SEER Pediatric Monograph
(ICCC group II) and germ cell (ICCC group
X) for 1986-94 (Figures 7-9). Among those
older than 9 years of age, there were very
low incidence rates for neuroblastoma
(ICCC group IVa), retinoblastoma (ICCC
group V), Wilms’ tumor (ICCC group VIa),
and hepatic tumors (ICCC group VII).
SURVIVAL
The cancer survival rate for children
has greatly improved over time. Even since
the mid-1970s there have been large im-
provements in short term and long term
survival (Figure 10). There were improve-
ments in survival for many forms of child-
hood cancer (Figure 11). The principal
reason for the gain for total childhood
cancer is due to the improvement in the
survival of leukemia, especially acute
lymphocytic leukemia, which includes
about a third of the pediatric cases. This is
due primarily to improvements resulting
from more efficacious chemotherapy agents.
RISK FACTORS
Throughout this monograph, there are
discussions of potential causes and risk
factors for individual childhood cancers.
The discussion below provides background
for considering the strength of the epide-
miological evidence available for each risk
factor. Since the evidence on risk factors
varies, each risk factor table has the factors
characterized by one of the following:
• Known risk factors: Most epidemi-
ologists consider these characteris-
tics or exposures to be ‘causes’ of the
particular cancer. The scientific
evidence meets all or most of the
criteria described earlier. However,
many individuals in the population
may have the characteristic or
%
%
%
%
%
%
%
%
%
%
%
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
1975 1980 1985 1990 1995
Year of diagnosis
0
10
20
30
40
50
60
70
80
90
100
Percent surviving
Survival rate:
1year from dx
3 yrs from dx
5 yrs from dx
10 yrs from dx
"
#
$
%
Figure 10: Trends in relative survival rates for all
childhood cancers, age <20, all races, both sexes
SEER (9 areas), 1975-94
%
%
%
%
%
$
$
$
$
$
'
'
'
'
'
&
&
&
&
&
!
!
!
!
!
1975-78 1979-82 1983-86 1987-90 1991-94
Year of diagnosis
0
20
40
60
80
100
Percent surviving
Leukemia
Lymphoma
Brain/CNS
Sympathetic Nerv.
Retinoblastoma
!
&
'
$
%
%
%
%
%
%
$
$
$
$
$
'
'
'
'
'
&
&
&
&
&
!
!
!
!
!
&
&
&
&
&
1975-78 1979-82 1983-86 1987-90 1991-94
Year of diagnosis
0
20
40
60
80
100
Percent surviving
Renal Hepatic Bone
Soft tissue Germ cell Carcinomas
& ! &
' $ %
Figure 11: Trends in 5-year relative cancer
survival rates by ICCC group, age <20
all races, both sexes, SEER (9 areas), 1975-94

INTRODUCTION
10
National Cancer Institute
SEER Pediatric Monograph
exposure and not develop cancer
because there are other contributory
factors.
• Suggestive but not conclusive evi-
dence: The scientific evidence link-
ing these characteristics or expo-
sures to the particular cancer meets
some but not all of the criteria
described earlier.
• Conflicting evidence: Some studies
show the putative risk factor to be
associated with higher risk but
others show no increased risk or
lower risk.
• Limited evidence: Very few studies
have investigated the putative risk
factor. The existing studies may
have investigated the exposure in a
superficial manner or methodologic
issues may make the results difficult
to interpret.
Finding causes of any disease is usu-
ally a long, slow process. Epidemiologists
find clues in one study that they follow-up
in later studies. Only some of the clues are
useful. Current studies are designed to
help us learn whether or not previously
identified clues are likely to lead us to the
causes of a particular cancer. No one study
is likely to prove that a particular exposure
definitely causes a particular cancer. No
single study nor even a large number of
epidemiologic studies will enable a parent
to know why his or her child developed
cancer. However, each well designed and
well executed study will bring us closer to
understanding the causes of these cancers
within populations of children.
Multifactorial etiology
We also do not expect that all children
with a particular cancer developed it for the
same reason. In other words, we do not
think that one exposure, behavior or ge-
netic trait explains all or even a majority of
instances of a particular cancer. Rather, we
expect that a number of exposures and
characteristics of children each contribute
to a proportion of instances of a particular
cancer.
No one factor determines whether an
individual will develop cancer, even if a
specific exposure explains a high proportion
of the occurrence of a specific cancer.
Rather, it is the interaction of many factors
that produces cancer. This concept is
referred to as the multiple causation or
multifactorial etiology. The factors involved
may be genetic, constitutional or behavioral
characteristics of the individual or factors
external to the individual. Among the
many types of factors that might play a role
are genetic, immune, dietary, occupational,
hormonal, viral, socioeconomic, lifestyle,
and other characteristics of the individual
and the biologic, social, or physical environ-
ment.
The concept of multiple causation has
direct implications for the interpretation of
research on the causes of cancer. Suppose
that combinations of laboratory and epide-
miologic studies have shown that exposure
to chemical X causes leukemia. We know
that other factors must play a role since not
all children who were exposed to chemical
X developed leukemia. Thus, there must be
other factors that determine which of the
children exposed to chemical X will develop
leukemia.
Associations versus causes
Frequently, newspapers and television
report that some chemical, dietary habit, or
household product is purported to increase
the risk of cancer. These news stories tell
us about associations between an exposure
and a cancer. In other words, more of the
people who developed cancer than those
without cancer had the exposure. However,
an association between an exposure and

INTRODUCTION
11
National Cancer Institute SEER Pediatric Monograph
cancer does not necessarily mean that the
exposure causes cancer.
As an example, suppose a case-control
study (see Technical Appendix) finds that
more of the mothers of children with acute
lymphoblastic leukemia (ALL) than moth-
ers of controls used medication Y during
pregnancy. It is possible that medication Y
causes ALL, but there are also other expla-
nations. It may be that mothers of children
with ALL were more accurate in their
reporting of medication use than the con-
trol mothers. Since mothers are asked in
these studies to recall their use of medica-
tion and other substances during a preg-
nancy 5 or 10 years in the past, it is certain
that their reporting is not completely
accurate. Mothers of children with cancer
have probably thought about their expo-
sures during the relevant pregnancies more
intently than control mothers in their
search for an explanation of their children’s
illness. Case mothers may remember short
episodes of medication use whereas control
mothers may have forgotten them. Differ-
ences in the level of recall between mothers
of cases and mothers of controls may be
real or may reflect less accurate recall of
either group of mothers. This type of
differential recall may lead to erroneous
results for either group; such differential
recall would lead to inaccurate or biased
results, a problem designated as recall bias.
A recall bias would lead to an association or
disassociation between the medication and
cancer which would not be causal but
spurious or false. Another explanation of
an association between the medication and
cancer is that medication Y is used to treat
a medical condition and that the condition
rather than the medication confers the risk.
Epidemiologists would say that the condi-
tion is a confounder of the observed associa-
tion between the medication and cancer.
How do epidemiologists decide whether
an association between an exposure and a
disease is one of cause and effect? The
methods and processes of epidemiology and
their limitations make it nearly impossible
for a single study to prove that an exposure
causes a disease. There must be a number
of studies that epidemiologists can evaluate
using a set of criteria. The criteria are
described briefly but the order in which
they are described does not signify relative
importance.
1. Other possible explanations of the
observed association must be ruled
out, such as the medical condition
rather than the medication. In
another example, if one investigates
an association between eating hot
dogs and developing a specific
cancer, one must determine whether
high dietary fat intake or infrequent
fruit eating explains the association
and rule out these factors before
concluding hot dog consumption is
related to risk.
2. Epidemiologists consider the
strength of the association, that is,
the relative risk (see Technical
Appendix). An exposure associated
with a ten-fold increase in risk is
more likely to be a true cause than
an exposure associated with a two-
fold increase.
3. The consistency of an association is
considered. An association observed
in many different studies in differ-
ent populations using different
study methods is likely to be true.
4. The observation of a dose-response
relationship between the exposure
and the disease increases confidence
that the exposure is really related to
the disease. In a dose-response
relationship, the risk of disease
increases or decreases as the
amount of the exposure increases or
decreases. For example, the rela-
tionship between cigarette smoking

INTRODUCTION
12
National Cancer Institute
SEER Pediatric Monograph
and lung cancer shows a dose-
response in that heavy smokers
have a higher risk than light smok-
ers.
5. The association must be temporally
correct meaning that we must be
sure that the exposure actually
preceded development of the disease.
For example, a study might report
that barbiturate use increased the
risk of brain tumors. However,
barbiturates are used to control
seizures, which are often an early
symptom of a brain tumor. There-
fore, it may not be clear if barbitu-
rate use actually preceded the
development of the brain tumor or if
barbiturates were used to treat an
early symptom before the brain
tumor was diagnosed.
6. A biologically plausible association
is more likely to be true than one
without other supporting evidence.
For example, we have more confi-
dence that chemical X causes brain
tumors in humans if it is known to
cause brain tumors in animals.
All or most of these six criteria must be
met before an association between a dis-
ease and an exposure is considered a causal
association.
Structure of monograph
This monograph consists of a chapter
for each of the principal types of pediatric
cancers as designated by the ICCC. The
ICCC designated group is also used as the
chapter number except for group XII which
is less than 6% of the total and is not
shown in a separate chapter. Each of these
chapters discusses incidence, mortality, and
survival rates of the patients, as well as
trends in these measures by demographic
characteristics. Risk factors are also de-
scribed. The estimated number of cases in
the US for 1998 is given in each chapter.
These numbers are based on the American
Cancer Society’s overall cancer estimate of
12,400 [1] cases and on the SEER site
distribution for 1990-95.
In addition, there are separate chap-
ters on children younger than 1 year of age,
adolescents, and incidence vs. mortality
trends. The monograph is also available
from the SEER home page under publica-
tions (http://www-seer.ims.nci.nih.gov). There
is a technical appendix at the end of this
chapter which defines terms used in the
Introduction and in other chapters; it also
provides more details on methods and data
sources.
TECHNICAL APPENDIX
Age-adjusted rate: An age-adjusted rate is a
weighted average of the age-specific cancer inci-
dence (or mortality) rates, where the weights are the
proportions of persons in the corresponding age
groups of a standard population. The potential
confounding effect of age is reduced when comparing
age-adjusted rates computed using the same
standard population. For this report, the 1970
United States standard million is used as the
standard in computing all age-adjusted rates. Since
rates of childhood cancer vary widely by 5-year age
group, age-adjustment was used for any age group
representing more than one 5-year grouping. Age-
adjustment was performed by 5-year age group and
weighted by the 1970 US standard million popula-
tion.
Age-specific rates: Age-specific rates are usually
presented as a rate per million. The numerator of
the rate is the number of cancer cases found in a
particular 5-year age group in a defined population
divided by the number of individuals in the same 5-
year age group in that population. In this publica-
tion, there are some rates by single year of age for
time periods around the Census. Population
estimates by single year of age, race, sex, and
geographic region are not generally available for
intercensal years. The rates by single year of age
are plotted at half years. For example, the rate for
children age 1 year is plotted at 1.5 years since they
are an average 1 1/2 years of age.
Case-control study: A case-control study is an
epidemiologic study in which a group of individuals
with a disease, the cases, are compared to a group of
individuals without the disease, the controls.

INTRODUCTION
13
National Cancer Institute SEER Pediatric Monograph
Exposures or characteristics that are more common
in the cases than in the controls may be causes of
the disease. Exposures or characteristics that are
equally common in the cases and controls cannot be
causes of the disease. Almost all studies of child-
hood cancer are case-control studies because this
type of study is very useful in studying relatively
uncommon diseases.
Cohort study: A cohort study is an epidemiologic
study in which the incidence of disease is compared
between a group of individuals with an exposure or
characteristic and a group without that exposure or
characteristic. For example, smokers and nonsmok-
ers are followed and the incidence of heart disease is
compared in the two groups. Or, the incidence of
breast cancer is compared in women with and
without a BRCA1 gene mutation. This type of study
is rarely feasible in investigating the etiology of
childhood cancer. Since childhood cancer is rare,
especially if we consider that each cancer should be
studied separately, huge numbers of children (a few
hundred thousand) would have to be followed to
determine which children developed cancer.
EAPC (Estimated Annual Percent Change): The
Estimated Annual Percent Change (EAPC) was
calculated by fitting a regression line to the natural
logarithm of the rates (r) using calendar year as a
regressor variable, i.e. y = mx + b where y = Ln r
and x = calendar year. The EAPC = 100*(e
m
- 1).
Testing the hypothesis that the Annual Percent
Change is equal to zero is equivalent to testing the
hypothesis that the slope of the line in the above
equation is equal to zero. The latter hypothesis is
tested using the t distribution of m/SE
m
with the
number of degrees of freedom equal to the number
of calendar years minus two. The standard error of
m, i.e. SE
m
, is obtained from the fit of the regression
[5]. This calculation assumes that the rates in-
creased/decreased at a constant rate over the entire
calendar year interval. The validity of this assump-
tion has not been assessed. In those few instances
where at least one of the rates was equal to zero, the
linear regression was not calculated. The differ-
ences between incidence and mortality trends for
the time period 1975-79 versus those for the most
recent five-year period are tested for statistical
significance using a t statistic with six degrees of
freedom defined as the difference in the regression
coefficients divided by the standard error of the
difference [5].
Follow-up: SEER areas attempt to follow-up all
cases till death. Although the overall proportion of
cancer patients of all ages who are lost to follow-up
is only about 5%, for pediatric cases (age 0-19) it is
much larger - about 14%. Since survival rates are
relatively high, follow-up can be difficult, especially
as the child gets older. When children leave their
parents’ home, they change addresses and, espe-
cially for females, they may change last names.
ICCC classification: At the time the World Health
Organization’s (WHO) International Agency for
Research on Cancer (IARC) published their first
monograph on Childhood Cancer [6] in 1988, Dr. R.
Marsden published an annex giving a classification
scheme for childhood cancer that consisted of 12
groups based chiefly on histologic type. The classifi-
cation by Marsden has been modified and is now
called the International Classification of Childhood
Cancers [4].
Incidence rate: The cancer incidence rate is the
number of new cancers of a specific site/type
occurring in a specified population during a year,
expressed as the number of cancers per one million
people. It should be noted that the numerator of the
rate can include multiple primary cancers occurring
in one individual. This rate can be computed for
each type of cancer as well as for all cancers com-
bined. Except for five-year age-specific rates, all
incidence rates are age-adjusted to the 1970 US
standard million population. Rates are for invasive
cancer only, unless otherwise specified.
Mortality data: The mortality data are from public
use files provided by the National Center for Health
Statistics (NCHS) and cover all deaths in the
United States. All mortality rates were based on
the underlying cause of death. The rates presented
for 1975-1978 were coded to the International
Classification of Diseases - 8
th
revision and for 1979
to 1995 to the ICD 9
th
revision [7]. Unfortunately
mortality of all specific groups of the ICCC pediatric
classification are not available from US mortality
files due to the fact that the codes used for coding
death certificates do not include such morphologic
types as neuroblastomas and retinoblastomas.
Certain groups can be identified as specific entities
on death certificates: Leukemias, Lymphomas,
Bones, Brain and other CNS tumors, and Hodgkin’s
and Non-Hodgkin’s lymphoma. However, such types
of cancer as Retinoblastomas, Germ cell tumors,
Wilms’ tumor, and certain carcinomas can not be
identified on death certificates. Even though
neuroblastomas are not coded separately, they were
coded to different groups in the ICD-8 and ICD-9.
For these analyses to make the data comparable
over time, deaths coded to sympathetic nervous
system in the 8th revision were combined with
adrenal in the 9th revision.
Mortality rate: The cancer mortality rate is the
number of deaths with cancer given as the underly-
ing cause of death occurring in a specified popula-
tion during a year, expressed as the number of
deaths due to cancer per one million people. This
rate can be computed for each type of cancer as well

INTRODUCTION
14
National Cancer Institute
SEER Pediatric Monograph
as for all cancers combined. Except for age-specific
rates, all mortality rates are age-adjusted to the
1970 US standard million population.
Population data: Population estimates are obtained
each year from the US Bureau of the Census at the
county level by five-year age group (0-4, 5-9,..., 85
and over), sex, and race (including white and black).
SEER areas make county estimates for each state
available on the SEER areas Home Page
(http://www-seer.ims.nci.nih.gov) for race (whites,
blacks, non-white), 5-year age group, sex, and year of
diagnosis (each year 1973 to 1995). Additional
estimates can be obtained from the US Census
Bureau Home Page.
US Bureau of the Census (BOC) population
estimates for Hawaii were altered according to
independent estimates developed from sample
survey data collected by the Health Surveillance
Program (HSP) of the Hawaii Department of
Health. For Hawaii, the all races and black popula-
tions are the same as those sent by the BOC.
Proportions of the population by different racial
groups from the HSP were used to generate esti-
mates for whites, etc. Since the HSP survey was for
all of Hawaii and not by county, population esti-
mates were not broken down by county. The white
population estimates for Hawaii provided by the
BOC are generally larger than those generated by
the HSP. Since whites in Hawaii account for less
than two percent of the total white population
represented by the SEER reporting areas, white
incidence rates for the entire SEER Program are not
noticeably affected. Procedures for calculating rates
by race for Hawaii are currently under review.
Primary site/histology coding: Originally data for
site and histologic type were coded by the Interna-
tional Classification of Diseases for Oncology (ICD-
O) [8], but in 1990, ICD-O was revised and repub-
lished as the International Classification of Diseases
for Oncology, 2nd Edition (ICD-O-2) [7]. SEER
areas began using ICD-O-2 for cases diagnosed in
1992 and machine converted all previous data to
ICD-O-2. Most data for Non-Hodgkin’s Lymphoma
(NHL) can be classified by the Working Formulation
(WF) based on a conversion from ICD-O-2.
Relative risk: Whether or not an exposure increases
the risk of cancer and how much it does is expressed
in a measure called relative risk. The relative risk
is the risk of disease in those with the exposure
divided by the risk of disease in those without the
exposure.
• Relative risk less than 1.0 - the exposure
appears to lower the risk of the disease. For
example, a relative risk of 0.75 for taking
vitamin X supplements indicates that those
who took vitamin X had a risk that was 75%
of that for individuals who did not take
vitamin X. Or, taking vitamin X lowered one’s
risk by 25%.
• Relative risk of 1.0 - the exposure does not
affect the risk of the disease; the risk is the
same in those with the exposure as in those
without the exposure.
• Relative risk greater than 1.0 - the exposure
appears to increase the risk of the disease.
For example, a relative risk of 3 for taking
medication Y indicates that those taking the
medication had a risk that was three times
that of those not taking the medication.
Relative survival rate: The relative survival rate is
calculated using a procedure described by Ederer,
Axtell, and Cutler [9] whereby the observed survival
rate is adjusted for expected mortality. The relative
survival rate represents the likelihood that a
patient will not die from causes associated specifi-
cally with their cancer at some specified time after
diagnosis. It is always larger than the observed
survival rate for the same group of patients.
Risk factor: A risk factor is a characteristic or
exposure that increases the risk of disease. A risk
factor might be exposure to high levels of radon,
having a diet low in vitamin A, having a family
history of colon cancer, or having a high cholesterol
level.
SEER Program: This program started in 1973, as
an outgrowth of the NCI’s Third National Cancer
Survey. NCI contracts out with various medically
oriented non-profit organizations, local city or state
Health Departments or Universities for collection of
these data. Contracts for collecting this data are
with the entire states of Connecticut, Iowa, New
Mexico, Utah, and Hawaii and with the metropoli-
tan areas of Los Angeles, California; Detroit,
Michigan; San Francisco-Oakland and San Jose-
Monterey, California; Seattle-Puget Sound, Wash-
ington; and Atlanta, Georgia. These organizations
collect data on all cancers except basal and squa-
mous cell skin cancers. Although data are collected
on all people having cancer, the material for this
study used children from birth through age 19 years.
Only residents of the areas designated above are
included so that the base populations can be
properly determined.

INTRODUCTION
15
National Cancer Institute SEER Pediatric Monograph
Reference List
1. Wingo P. American Cancer Society. Personal
communication based on unpublished data
from Cancer facts and figures, 1998. Atlanta,
1998.
2. Ries LAG, Kosary CL, Hankey BF, Miller BA,
Clegg L, Edwards BK (eds). SEER Cancer
Statistics Review 1973-1995, National Cancer
Institute, http://www.seer.ims.nci.nih.gov, 1998.
3. Percy C, Van Holten V, and Muir C, Eds.
International classification of diseases for
oncology, Second Ed.,World Health Organiza-
tion, Geneva, 1990.
4. Kramarova E and Stiller CA, The International
Classification of Childhood Cancer, Int J
Cancer. 68:759-765, 1996.
5. Kleinbaum DG, Kupper LL, Muller KE.
Applied Regression Analysis and Other
Multivariable Methods. North Scituate,
Massachusetts: Duxbury Press, 1988: 266-268.
6. Parkin DM, Stiller CA, Draper GJ, Bieber CA,
Terracini B, and Young JL (eds). International
incidence of childhood cancer, WHO, IARC
Scientific Publication No.87, Lyon, 1988.
7. World Health Organization, International
Classification of Diseases, 1975 Revision, vols.
1 and 2, Geneva, 1977.
8. World Health Organization, International
Classification of Diseasess for Oncology, First
Edition, Geneva, 1976.
9. Ederer F, Axtell LM, Cutler SJ. The relative
survival rate: A statistical methodology. Natl
Cancer Inst Monogr 1961; 6:101-121.

16
National Cancer Institute
SEER Pediatric Monograph

ICCC ILEUKEMIA
17
National Cancer Institute
SEER Pediatric Monograph
Malcolm A. Smith, Lynn A. Gloeckler Ries, James G. Gurney, Julie A. Ross
HIGHLIGHTS
Incidence
♦ For the years from 1990-95, the leukemias represented 31% of all cancer cases
occurring among children younger than 15 years of age and 25% of cancer cases
occurring among those younger than 20 years of age. In the US there are approxi-
mately 3,250 children diagnosed each year with leukemia and 2,400 with acute
lymphoblastic leukemia (ALL).
♦ The relative contribution of leukemia to the total childhood cancer burden varies
markedly with age, being 17% in the first year of life, increasing to 46% for 2 and 3
year olds, and then decreasing to only 9% for 19 year olds (Figure I.1).
♦ The two major types of leukemia were ALL comprising nearly three-fourths and
acute non-lymphocytic comprising 19%.
♦ There was a sharp peak in ALL incidence among 2-3 year olds (> 80 per million)
which decreases to a rate of 20 per million for 8-10 year olds. The incidence of ALL
among 2-3 year olds is approximately 4-fold greater than that for infants and is
nearly 10-fold greater than that for 19 year olds (Figure I.2a).
♦ Leukemia rates are substantially higher for white children than for black children,
with rates of 45.6 versus 27.8 per million for the period from 1986-95 for children
0-14 years old (Table I.4). This difference between white and black children is
most apparent when examining rates of leukemia by single year of age (Figure
I.3), with a nearly 3-fold higher incidence at 2-3 years of age for white children
compared to black children.
♦ The incidence of leukemia among children younger than 15 years of age has
shown a moderate increase in the past 20 years (Figure I.4) with the trend prima-
rily reflecting an increase in ALL incidence during this period. The rates of leuke-
mias other than ALL did not appear to increase from 1977 to 1995 (Figure I.5)
Survival
♦ Survival for children with ALL has markedly improved since the early 1970s, and
overall survival for all children with ALL is now approximately 80% (Figure I.8).
A number of improvements in treatment during this period have undoubtedly
contributed to the improved survival.
♦ Survival for children with ALL is very dependent upon age at diagnosis, with the
most favorable outcome observed for children older than 1 year of age and younger
than 10 years of age.
Risk factors
♦ With the exception of prenatal exposure to x-rays and specific genetic syndromes,
little is known about the causes of childhood ALL (Table I.5).
♦ Different risk factors are emerging for childhood AML that distinguish the disease
from ALL, and this may provide avenues for future epidemiological studies (Table
I.6).

ICCC I
18
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
INTRODUCTION
The leukemias of childhood are cancers
of the hematopoietic system, involving in
most cases, malignant transformation of
lymphoid progenitor cells [1] and less
commonly transformation of myeloid pro-
genitor cells [2]. The leukemias account for
the largest number of cases of childhood
cancer and are the primary cause of cancer
related mortality of children in the United
States. Approximately 3,250 children and
adolescents younger than 20 years of age
are diagnosed with leukemia each year in
the US, of which 2,400 are acute lympho-
blastic leukemia. For the years from 1986-
94, the leukemias represented 32% of all
cancer cases occurring among children
younger than 15 years of age and 26% of
cancer cases occurring among those
younger than 20 years of age. However,
the relative contribution of leukemia to the
total childhood cancer burden varied mark-
edly with age, being 17% in the first year of
life, increasing to 46% for 2 and 3 year olds,
and then decreasing to only 9% for 19 year
olds. To further illustrate the contribution
Figure I.1 gives the incidence rates for both
leukemia and total cancer (the sum of
leukemia and non-leukemia) by single year
of age.
1
This chapter focuses on the following
topics related to the incidence of leukemia
among children in the United States: (1)
the relative frequencies of the leukemia
subtypes that occur among children; (2)
variation in the incidence of the specific
types of leukemia by age; (3) differences in
incidence between males and females; (4)
differences in incidence between white and
black children; and (5) variation in leuke-
mia incidence over time. In terms of sur-
vival for children with leukemia, the chap-
ter focuses on three primary topics: (1)
comparison of survival rates for children
with ALL and AML; (2) the impact of age at
diagnosis on survival; and (3) the remark-
Figure I.1: Total childhood cancer age-specific incidence rates
by leukemia versus non-leukemia, all races, both sexes, SEER, 1986-94
45
60
96
94
73
52
42
38
29
24
26
27
25 25
26
27 27
25 25
20
224
131
118
112
87
83
74
74
68
70 68
80
95
108
118
136
167
178
203 208
< 1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Age (in years) at diagnosis
0
50
100
150
200
250
300
Average annual rate per million
Leukemia
Non-Leukemia
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.

ICCC ILEUKEMIA
19
National Cancer Institute SEER Pediatric Monograph
Table I.1: Percent distribution within ICCC subcategories for leukemia and age-adjusted*
incidence rates for specific ICD-O codes, age <20, all races, both sexes, SEER, 1975-95
Diagnostic Group Specific Diagnosis: Rate per million % of Cases
Ia: Lymphoid leukemia 29.2 100.0%
9820: Lymphoid leukemia, NOS 0.2%
9821: Acute lymphoblastic 99.2%
9822: Subacute lymphoid 0.0%
9823: Chronic lymphocytic 0.1%
9824: Aleukemic lymphoid 0.2%
9825: Prolymphocytic leukemia 0.1%
9826: Burkitt's cell leukemia 0.5%
9827: Adult T-cell 0.0%
9850: Lymphosarcoma cell 0.0%
Ib: Acute non-lymphocytic 7.6 100.0%
9840: Erythroleukemia 0.4%
9841: Acute erythremia 0.2%
9861: Acute myeloid leukemia 68.7%
9864: Aleukemic myeloid 0.0%
9866: Acute promyelocytic 7.1%
9867: Acute myelomonocytic 9.3%
9891: Acute monocytic leukemia 9.1%
9894: Aleukemic monocytic 0.0%
9910: Acute megakaryoblastic 5.1%
Ic: Chronic myeloid leukemia 1.3 100.0%
9863: Chronic myeloid leukemia 98.6%
9868: Chronic myelomonocytic 1.4%
Id: Other specified leukemias 0.2 100.0%
9830: Plasma cell leukemia 0.0%
9842: Chronic erythremia 0.0%
9860: Myeloid leukemia, NOS 33.3%
9862: Subacute myeloid 0.0%
9870: Basophilic leukemia 0.0%
9880: Eosinophilic leukemia 0.0%
9890: Monocytic leukemia, NOS 8.3%
9892: Subacute monocytic 0.0%
9893: Chronic monocytic 0.0%
9900: Mast cell leukemia 0.0%
9930: Myeloid sarcoma 58.3%
9931: Acute panmyelosis 0.0%
9932: Acute myelofibrosis 0.0%
9940: Hairy cell leukemia 0.0%
9941: Leukemia 0.0%
Ie: Unspecified leukemias 1.2 100.0%
9800: Leukemia, NOS 20.5%
9801: Acute leukemia, NOS 79.5%
9802: Subacute leukemia, NOS 0.0%
9803: Chronic leukemia, NOS 0.0%
9804: Aleukemia leukemia, NOS 0.0%
*Adjusted to the 1970 US standard population

ICCC I
20
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
able improvements in survival rates for
children with ALL during the past 20
years.
Classification system
Before discussing topics related to
childhood leukemia incidence and outcome,
it is necessary to describe the specific
diagnoses that are included among the
Diagnostic Groups for leukemia of the
International Classification of Childhood
Cancer (ICCC). Table I.1 illustrates that
acute lymphoblastic leukemia (ALL) ac-
counted for approximately 99% of cases
among the lymphoid leukemia (Ia) diagnos-
tic group, so that this ICCC diagnostic
group is essentially synonymous with ALL.
The “acute non-lymphocytic leukemia”
diagnostic category Ib is henceforth re-
ferred to as the acute myeloid leukemia
(AML) category since this is the preferred
terminology [3], and it encompasses the
various subtypes of AML that occur in
children. The other three ICCC diagnostic
categories combined accounted for only 6-
7% of total leukemia cases in children. The
chronic myeloid leukemias diagnostic group
(Ic) included approximately 3% of leukemia
cases occurring in the younger than 20
years of age group during the period from
1990-95, while the “other specified leuke-
mia” diagnostic group (Id) included fewer
than 1% of the leukemia cases. Approxi-
mately 3% of leukemia cases were included
in the unspecified leukemia category (Ie)
for the period from 1990-95.
INCIDENCE
Age-specific incidence
Table I.2 shows the incidence and
relative proportion of specific diagnostic
categories by 5-year age groups. For the
younger than 15 years of age, ALL repre-
sented 78% of leukemia cases, while the
AML subgroup (Ib) represented 16% of
cases. The relative frequency of AML
increased in the second decade of life as
that of ALL decreased. While AML repre-
sented only 13-14% of leukemia cases in
the first 10 years of life, it accounted for
36% of leukemia cases among 15-19 year
olds. The incidence and relative contribu-
tion of the chronic myeloid leukemias also
increased with age, representing about 9%
of cases among 15-19 year olds.
As is apparent from Table I.2, the
incidence of leukemia among children
varied considerably with age. Figure I.2a
illustrates that this variation was the
result of a sharp peak in ALL incidence
among 2-3 year old children (incidence over
80 per million), which returned to a rate of
20 per million for 8-10 year old children.
The incidence of ALL among 2-3 year old
Table I.2: Age-adjusted incidence rates per million for specific leukemia by age groups
all races, both sexes, SEER, 1990-95
Age (in years) at diagnosis <5 5-9 10-14 15-19 <15*
Total leukemia 72.4
(100%)
38.0
(100%)
25.9
(100%)
26.0
(100%)
43.8
(100%)
ALL
58.1
(80%)
30.6
(81%)
17.4
(67%)
13.0
(50%)
34.0
(78%)
AML (Ib)
10.3
(14%)
5.0
(13%)
6.2
(24%)
9.3
(36%)
7.0
(16%)
CML (Ic) 1.1
(2%)
0.7
(2%)
1.1
(4%)
2.2
(9%)
1.0
(2%)
Other specified leukemias
(Id)
0.3
(-)
0.3
(1%)
0.1
(-)
0.1
(-)
0.2
(1%)
Unspecified leukemias (Ie)
2.2
(3%)
1.0
(3%)
0.6
(2%)
1.1
(4%)
1.2
(3%)
* Rates are adjusted to the 1970 US standard population. Numbers in parentheses represent the
percentage of the total cases for the specific age group.

ICCC ILEUKEMIA
21
National Cancer Institute SEER Pediatric Monograph
children was approximately 4-fold greater
than that for infants and was nearly 10-
fold greater than that for 19 year olds. The
distinctive shape of the age-incidence curve
for ALL can be well-described by a math-
ematical model which assumes that child-
hood ALL results from two events required
for full malignant transformation with the
first of these events occurring in utero [4].
Experimental confirmation has been ob-
tained for the initiation of childhood ALL in
utero [5,6].
The incidence of AML in children also
varied with age (Figure I.2b), but with a
different pattern than that for ALL. AML
rates were highest in the first 2 years of
life, but subsequently decreased with a
nadir at approximately 9 years of age,
followed by slowly increasing rates during
the adolescent years. The incidence of
leukemia cases in the “chronic myeloid
leukemia” category (Ic) likewise showed
substantial variation with age. As shown
in Figure I.2c, there was a peak in inci-
Figure I.2a: ALL (Ia): 1986-94, and AML (Ib): 1976-84
and 1986-94 age-specific incidence rates, all races
both sexes, SEER
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
50
60
70
80
90
100
Average annual rate per million
ALL
AML
&
(
Figure I.2b: AML (Ib) age-specific incidence rates, all races
both sexes, SEER, 1976-84 and 1986-94 combined
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
2
4
6
8
10
12
14
Average annual rate per million
AML
(
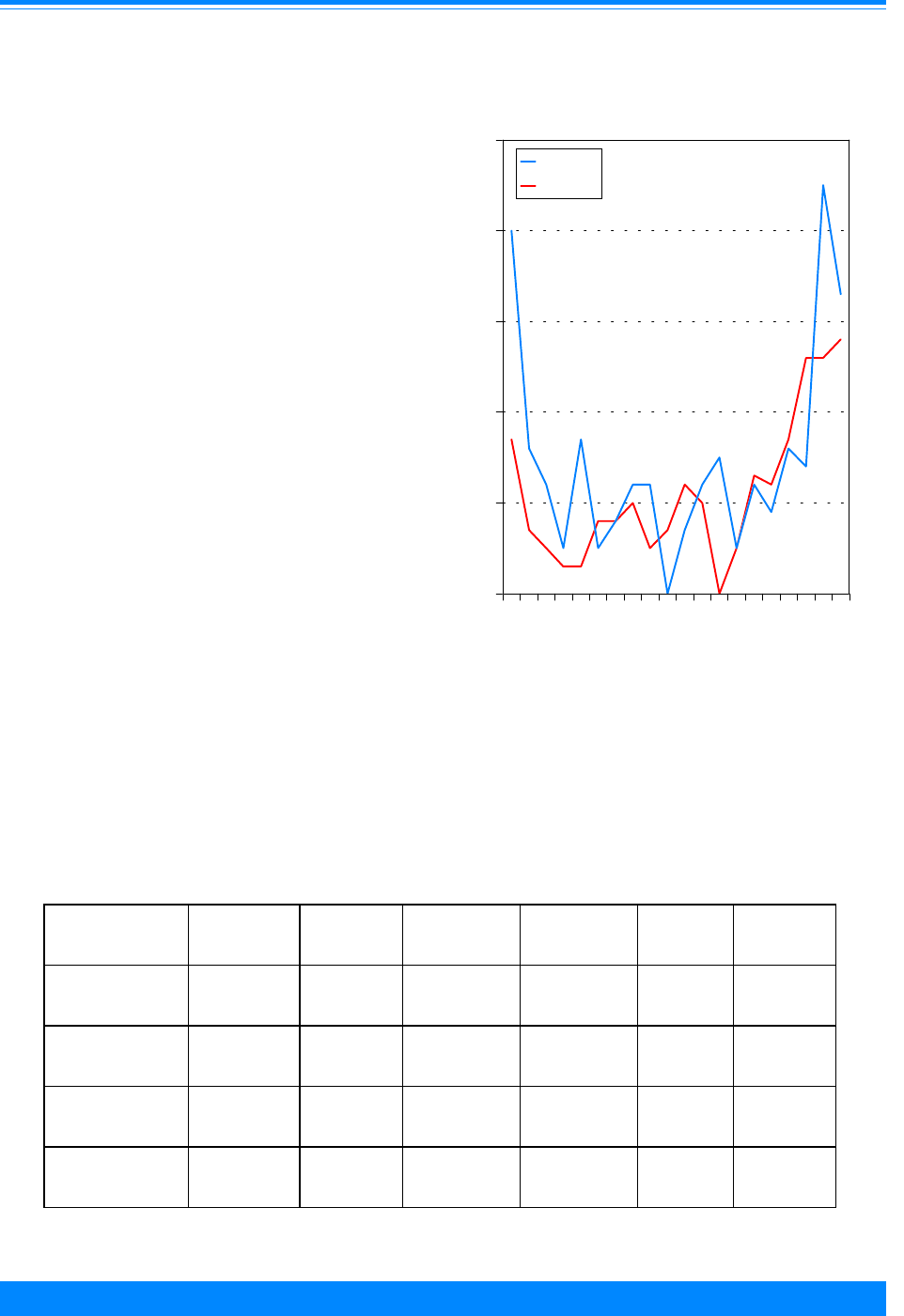
ICCC I
22
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
dence for males in the first year of life, with
subsequent lower rates for both males and
females until the late teen years. The
increase in CML incidence in the later teen
years appeared to represent the early
portion of the increasing incidence curve for
adult-type CML [7]. On the other hand, the
cases coded as “chronic myeloid leukemia”
in the first few years of life almost certainly
reflect cases of juvenile myelomonocytic
leukemia (previously termed juvenile
chronic myeloid leukemia), a diagnosis
associated primarily with young males
[8,9].
Sex-specific incidence
Table I.3 illustrates the incidence of
the various leukemia types separately for
males and females for the years 1990-95.
The incidence of ALL among children
younger than 15 years of age was consis-
tently higher among males (approximately
20%) relative to females. For the 15-19
year olds, however, the male preponderance
was greater, with males having a 2-fold
higher ALL incidence than females. The
incidence of AML was similar for males and
females for all age groups. For the CML
category, there was a 4-fold higher rate for
males than females for the younger than 5-
year age group, a difference that was not
present for older age groups. As noted
above, these cases likely represent juvenile
myelomonocytic leukemia, which has a
known male predominance [9].
Table I.3: Male to female ratios of age-adjusted leukemia incidence rates per million by
type and age group, all races, SEER, 1990-95
<5 Yrs
Rate
M/F Ratio
5-9 Yrs
Rate
M/F Ratio
10-14 Yrs
Rate
M/F Ratio
15-19 Yrs
Rate
M/F Ratio
<15 Yrs
Rate*
M/F Ratio
<20 Yrs
Rate*
M/F Ratio
Total leukemia
M = 78.5
F = 65.9
M/F = 1.2
M = 40.3
F = 35.7
M/F = 1.1
M = 28.4
F = 23.1
M/F = 1.2
M = 28.4
F = 23.4
M/F = 1.2
M = 47.4
F = 40.1
M/F = 1.2
M = 42.7
F = 36.0
M/F = 1.2
ALL M = 63.7
F = 52.3
M/F = 1.2
M = 32.2
F = 29.0
M/F = 1.1
M = 18.8
F = 16.0
M/F = 1.2
M = 17.2
F = 8.6
M/F = 2.0
M = 36.7
F = 31.2
M/F = 1.2
M = 31.9
F = 25.6
M/F = 1.2
AML (Ib) M = 10.0
F = 10.6
M/F = 0.9
M = 5.8
F = 4.3
M/F = 1.3
M = 6.7
F = 5.7
M/F = 1.2
M = 8.3
F = 10.4
M/F = 0.8
M = 7.4
F = 6.7
M/F = 1.1
M = 7.6
F = 7.6
M/F = 1.0
Chronic myeloid
leukemia (Ic)
M = 1.7
F = 0.4
M/F = 4.3
M = 0.4
F = 1.0
M/F = 0.4
M = 1.4
F = 0.9
M/F = 1.6
M = 1.6
F = 2.9
M/F = 0.6
M = 1.1
F = 0.8
M/F = 1.4
M = 1.3
F = 1.3
M/F = 1.0
*Adjusted to the 1970 US standard population
Figure I.2c: Chronic myeloid leukemia age-specific
incidence rates, all races, both sexes
SEER, 1976-84 and 1986-94 combined
)
)
)
)
))
))
)
)
)
)
)
)
)
)
)
)
))
"
"
"
"
"
"
"
"
""
"
"
"
"
"
"
"
"
"
"
0 1 2 3 4 5 6 7 8 9 1011 121314151617 181920
Age (in years) at diagnosis
0
1
2
3
4
5
Average annual rate per million
Male
Female
"
)

ICCC ILEUKEMIA
23
National Cancer Institute SEER Pediatric Monograph
Figure I.3: Leukemia age-specific incidence rates for white (1986-94)
and black (1976-84 and 1986-94) children, SEER
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
Average annual rate per million
White
Black
+
&
Black-white differences in incidence
Leukemia rates were substantially
higher for white children younger than 15
years of age compared to black children,
with rates of 45.6 versus 27.8 per million
for the period from 1986-95 (Table I.4).
This difference between white children and
black children was most apparent when
examining rates of leukemia by single year
of age (Figure I.3), with a nearly 3-fold
higher incidence at 2-3 years of age for
white children compared to black children.
The difference in leukemia incidence was
primarily the result of lower ALL rates
among black children (Table I.4), with ALL
incidence for white children younger than 5
years of age being more than twice that for
black children (63.2 versus 26.9 per mil-
lion). A lower ALL incidence for black
children was observed for each 5-year age
group up to 20 years of age. The incidence
of AML, unlike that for ALL, was similar
for white and black children for all age
groups (Table I.4).
Table I.4: White to black ratios of age-adjusted leukemia incidence rates per million by
type and age group, both sexes, SEER, 1986-95
<5 Yrs
Rate*
W/B Ratio
5-9 Yrs
Rate
W/B Ratio
10-14 Yrs
Rate
W/B Ratio
15-19 Yrs
Rate
W/B Ratio
<15 Yrs
Rate*
W/B Ratio
<20 Yrs
Rate*
W/B Ratio
Total
Leukemia
W = 77.2
B = 38.4
W/B = 2.0
W = 37.5
B = 28.6
W/B = 1.3
W = 27.4
B = 18.4
W/B = 1.5
W = 26.5
B = 15.2
W/B = 1.7
W = 45.6
B = 27.8
W/B = 1.6
W = 40.9
B = 24.7
W/B = 1.7
ALL W = 63.2
B = 26.9
W/B = 2.4
W = 31.8
B = 1.8
W/B = 1.8
W = 19.8
B = 11.6
W/B = 1.7
W = 14.3
B = 6.4
W/B = 2.2
W = 36.8
B = 18.3
W/B = 2.0
W = 31.2
B = 15.4
W/B = 2.0
AML (Ib)
W = 10
B = 7.7
W/B = 1.3
W = 3.8
B = 6.1
W/B = 0.6
W = 5.3
B = 5.1
W/B = 1.0
W = 8.3
B = 7.1
W/B = 1.2
W = 6.2
B = 6.2
W/B = 1.0
W = 6.7
B = 6.4
W/B = 1.1
*Adjusted to the 1970 U.S. standard population

ICCC I
24
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
TRENDS
The incidence of leukemia among
children younger than 15 years of age
increased in the past 20 years, as shown in
Figure I.4a. The estimated annual percent-
age change (EAPC) for total leukemia for
the period from 1977 to 1995 was 0.9% per
year, with the trend primarily reflecting an
increase in ALL incidence during this
period (EAPC for ALL, 0.9%). The rates of
leukemias, other than ALL, did not in-
crease significantly from 1977 to 1995
(Figure I.5), although the small number of
cases diagnosed each year for the less
common leukemia types results in consider-
able scatter in year to year rates, which
makes interpretations of trends difficult.
The higher rate of nonspecific classification
of leukemia cases (ICCC Category Ie) in the
years prior to 1977 (greater than 5 per
million in 1973 and 1974, but 1-2 per
million after 1977), is the reason for re-
stricting examinations of trends over time
for specific leukemia diagnoses to the
period from 1977 to 1995 [10].
While a model based on a constantly
increasing rate can be applied to the ALL
and the leukemia incidence data to esti-
mate an EAPC, visual inspection of the
incidence of ALL and total leukemia from
1977 to 1995 suggests that reality is more
complicated (Figure I.4a). For example,
ALL incidence for the 9 SEER areas and
the Los Angeles area combined peaked in
1989, and rates have been 5-10% below this
peak value in subsequent years. The
situation is further complicated by different
time trend patterns for ALL for the 9 SEER
areas compared with the Los Angeles area.
For the 9 SEER areas, ALL incidence has
been more or less stable since 1984. On the
other hand, ALL incidence for the Los
Angeles area showed more variability,
being higher in the late 1970s and early
Figure I.4b: Trends in ALL and non-ALL
age-adjusted* incidence rates, age <15
all races, both sexes, SEER, 1977-95
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
1977 1980 1983 1986 1989 1992 1995
Year of diagnosis
0
10
20
30
40
50
Average annual rate per million
ALL (9 SEER)
ALL (LA)
Non-ALL (LA)
Non-ALL (9 SEER)
&
#
'
,
*Adjusted to the 1970 US standard population
Figure I.4a: Trends in leukemia and ALL
age-adjusted* incidence rates, age <15
all races, both sexes, SEER, 1977-95
)
))
)
)
)
))
)
)
)
)
)
)
)
)
)
)
)
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
1977 1980 1983 1986 1989 1992 1995
Year of diagnosis
0
10
20
30
40
50
Average annual rate per million
Total Leukemia
ALL
Non-ALL
$
&
)
*Adjusted to the 1970 US standard population
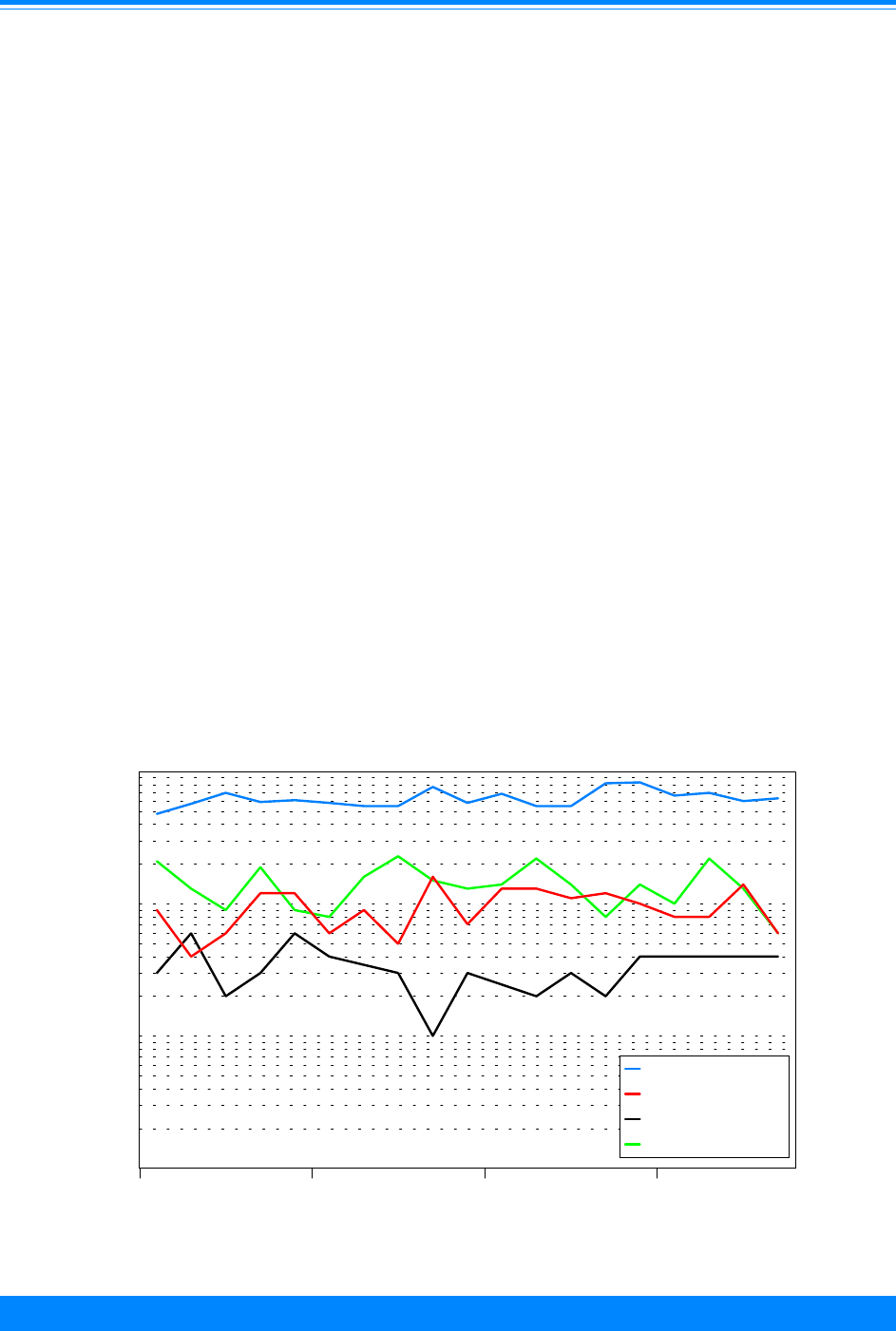
ICCC ILEUKEMIA
25
National Cancer Institute SEER Pediatric Monograph
1980s, then decreasing to a nadir in 1984-
85, and subsequently increasing to rates
higher than those for the 9 SEER areas
(Figure I.4b). For 1990 to 1995, ALL rates
can be calculated for Hispanic and non-
Hispanic children. For non-Hispanic chil-
dren, the ALL rates were similar between
Los Angeles (LA) and the 9 SEER areas.
However, the ALL rates for Hispanic chil-
dren were higher than those for non-His-
panic children. Since over one-half of the
children younger than 15 residing in Los
Angeles were Hispanic, a much higher
proportion than for the 9 SEER areas, the
overall higher ALL rates in Los Angeles can
be explained by the higher proportion of
Hispanic children living in LA. In addition,
between 1990 and 1995, there were in-
creases in the population of Hispanic chil-
dren under 15 and decreases in the popula-
tion of non-Hispanic children under 15 in
LA. This change in population characteris-
tics would tend to produce increased rates
of ALL due to the higher rates of ALL
among Hispanic children. Therefore, the
higher ALL rates in LA can be explained by
the higher proportion of Hispanic children
in LA, and the increase in ALL rates for LA
can be at least partially explained by
increases in the percentage of Hispanic
children in LA. Ongoing monitoring of
childhood leukemia trends, as well as
epidemiologic and basic laboratory studies,
are needed to develop a better understand-
ing of the pathogenesis of childhood leuke-
mia and an enhanced ability to explain
changes in leukemia incidence over time.
Figure I.6 illustrates the incidence of
ALL for white and black children for the
period from 1977 to 1995. While the inci-
dence of ALL for white children increased
at an overall rate of approximately 1% per
year since 1977, there was no apparent
increase in ALL rates for black children
during this same period.
Figure I.5: Trends in non-ALL leukemia age-adjusted* incidence rates
age <15, all races, both sexes, SEER, 1977-95
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
%
%
%
%
%
%
%
%
%
%
%
%
%
%%
+
+
+
+
++
+
+
+
+
+
++
+
+
+
++
+
$
$
$
$
$
$
$
$$
$
$
$
$$
$
$
$
$
$
1977 1982 1987 1992
Year of diagnosis
0.01
0.1
1
10
Average annual rate per million (log scale)
IB (ANLL)
IC (CML)
ID (Other)
IE (Unspecified)
$
+
%
&
*Adjusted to the 1970 US standard population

ICCC I
26
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
Figure I.7 illustrates the variation in
ALL incidence for white children for specific
5-year age groups. Incidence rates for
white children 0-4, 5-9, 10-14, and 15-19
years of age demonstrated modest increases
when the entire time period was considered.
Because the incidence of ALL was greater
for those younger than 5 years of age than
for the older age groups, the increasing
rates for those younger than 5 years of age
accounted for the largest proportion of the
overall increase in ALL rates for white
children.
SURVIVAL
Survival for children with ALL mark-
edly improved from 1975-84 to 1985-94,
with 5-year survival rates for children
younger than 20 years of age increasing
from 61% to 77% (Figure I.8). A number of
improvements in treatment during this
period have undoubtedly contributed to the
improved survival rate, including: a) identi-
fication of increasingly effective methods of
central nervous system prophylaxis [1]; b)
identification of the contribution of treat-
ment intensification to improved outcome
for selected groups of patients [27-29].
Examples of effective methods of treatment
intensification include use of post-induction
consolidation with high-dose methotrexate
[28, 29] and use of post-remission re-induc-
tion/re-consolidation regimens (“delayed
intensification”) [27].
Survival for children with ALL is very
dependent upon age at diagnosis. For the
years 1985-94, 5-year survival rates were
highest for the 1-4 year age group and the
5-9 year age group (85% and 80%, respec-
tively) (Figure I.8). Infants had the poorest
outcome (37% 5-year survival rate), fol-
Figure I.7: Trends in ALL age-specific incidence
rates by year of diagnosis, white children
both sexes, SEER, 1977-95
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
+
+
++
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
1977 1982 1987 1992
Year of diagnosis
0
20
40
60
80
Average annual rate per million
<5 Years 5-9 Years
10-14 Years 15-19 Years
& )
+ %
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
+
+
+
+
++
+
++
+
+
+
+
+
+
+
+
+
+
1977 1982 1987 1992
Year of diagnosis
0
10
20
30
40
50
Average annual rate per million
White
Black
+
)
*Adjusted to the 1970 US standard population
Figure I.6: Trends in ALL age-adjusted*
incidence rates by race, age <15
both sexes, SEER, 1977-95
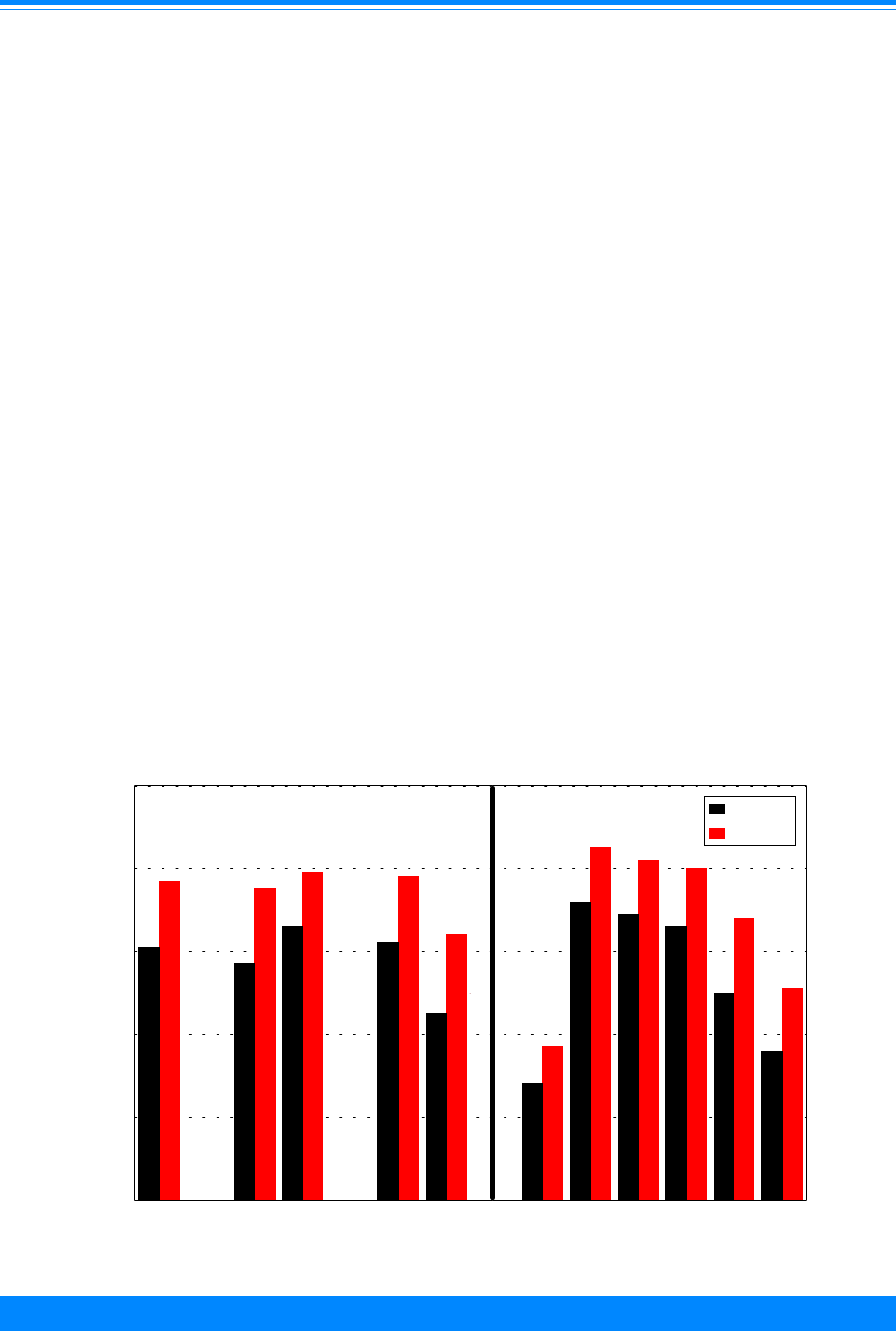
ICCC ILEUKEMIA
27
National Cancer Institute SEER Pediatric Monograph
lowed by the 15-19 year age group (51% 5-
year survival rate). The favorable progno-
sis of 1-9 year old children is likely related
to the relatively high proportion of cases in
this age range with favorable biological
subtypes (e.g., cases with hyperdiploid DNA
content or with the TEL-AML1 gene rear-
rangement) [30-32]. The poor prognosis for
infants with ALL reflects the high fre-
quency of cases with rearrangements of the
MLL gene on chromosome band 11q23
[33,34]. The less favorable outcome for
adolescents and young adults is likely due
in part to the increased relative frequency
of higher risk ALL subtypes (e.g., Philadel-
phia chromosome positive ALL and T-cell
ALL).
For the younger than 20 year old
population, 5-year survival rates were
slightly higher for females than for males
(79% versus 75%) (Figure I.8). Five-year
survival rates for black children younger
than 20 years of age with ALL were lower
than for white children (64% versus 78%).
While the poorer outcome for black children
with ALL could represent differences
between black and white children in the
pharmacokinetics or pharmacodynamics of
the drugs used for ALL treatment or differ-
ences in access to health care, the relative
paucity among black children of the most
curable ALL subtypes that occur at higher
incidence among white children younger
than 10 years of age may also contribute to
the poorer outcome observed for black
children.
Survival for children with AML was
substantially lower than that for children
with ALL (Figure I.9). While outcome for
children with AML improved significantly
from 1975-84 to 1985-94, 5-year survival
rates were only 41% for the period 1985-94
for the younger than 20 year old age group.
In contrast to ALL, older children and
adolescents with AML had outcome that
was similar to that observed for the
Figure I.8: ALL 5-year relative survival rates by sex, race, age and
time period, SEER (9 areas), 1975-84 and 1985-94
61
57
66
62
45
28
72
69
66
50
36
77
75
79
78
64
37
85
82
80
68
51
Total Male Female White Black <1 1-4 <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex Race Age
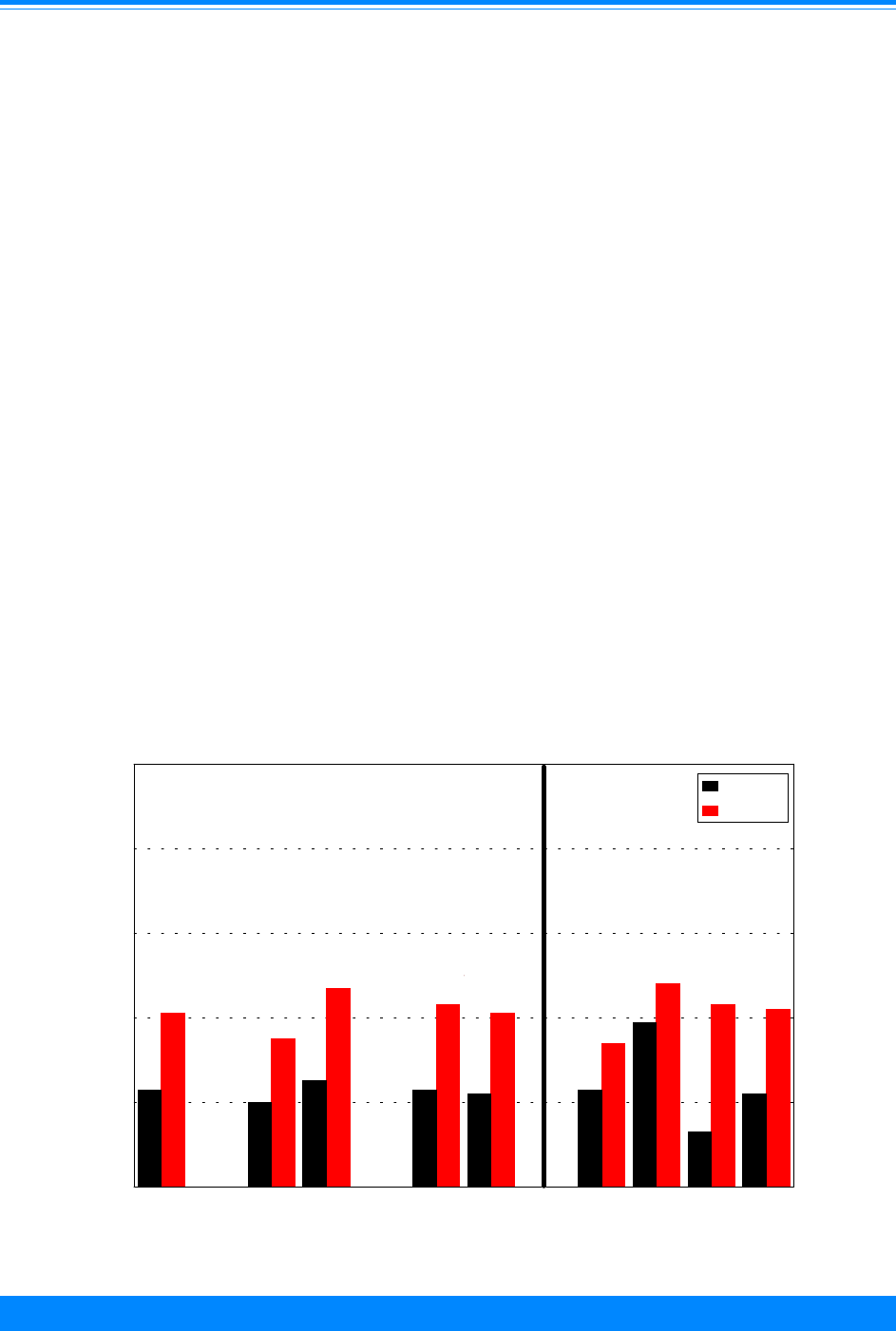
ICCC I
28
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
younger than 5-year age group, with the
younger than 5-year age group having
somewhat lower outcome than for the older
age groups. As was the case for ALL,
outcome for females with AML was some-
what better than outcome for males. In
contrast to the poorer outcome for black
children with ALL, for AML outcome was
similar for white and for black children
younger than 20 years of age.
RISK FACTORS
Tables I.5 and I.6 summarize current
knowledge of the causes of childhood ALL
and AML. With the exception of prenatal
exposure to x-rays and specific genetic
syndromes, little is known about the causes
of and risk factors for childhood ALL [13].
It is important to note that ALL is a hetero-
geneous grouping of biological subtypes of
leukemia, and smaller studies of the past
may have lacked sufficient statistical power
to examine potential risk factors. Thus, one
emerging theme concerning the etiology of
childhood ALL is the need to separately
study different biological groups of ALL.
For example, the cases of ALL that arise in
infants and that have rearrangements of
the MLL gene on chromosome 11 appear to
have different epidemiological associations
than cases that arise in young children that
typically have B-precursor ALL
immunophenotype and hyperdiploid DNA
content [14-16]. Recognition of the need to
study these different ALL subtypes inde-
pendently has been one impetus for larger
studies of the etiology of ALL. In these
larger studies some intriguing associations
have emerged that can be followed up
further in more focused investigations. For
example, high birth weight and maternal
history of fetal loss have been associated
primarily with ALL occurring in children
younger than 2 years of age [17-19].
From a global perspective, childhood
ALL appears to be much more common in
Figure I.9: AML 5-year relative survival rates by sex, race, age and
time period, SEER (9 areas), 1975-84 and 1985-94
23
20
25
23
22
23
39
13
22
41
35
47
43
41
34
48
43
42
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
ICCC ILEUKEMIA
29
National Cancer Institute SEER Pediatric Monograph
countries of the developed world than in
those of the developing world, a difference
that has been attributed to different pat-
terns of exposure of children in these popu-
lations to infectious agents [20,21]. A
delayed pattern of infections, as found in
countries such as the United States, is
hypothesized to somehow cause (or allow)
ALL to occasionally develop, possibly
through alterations in immune function.
However, in the absence of an understand-
ing of specific infectious agents or specific
immune perturbations associated with the
pathogenesis of ALL, it is not possible to
apply these theories to explain the trends
in incidence for childhood ALL observed in
the United States since the 1970s.
Table I.5: Current knowledge on causes of acute lymphoblastic leukemia (ALL)
Exposure or Characteristic Comments References
Sex Overall, there is about a 30% higher incidence in males
compared to females.
16,22,36
Age There is a peak in the incidence between the ages of about
2 and 5.
16,22,36
Race There is an approximate 2-fold higher risk in white
children compared to black children.
16,22,36
Higher socioeconomic status Increased risk has been fairly consistently associated with
the most common ALL (diagnosed at ages 2 - 5 years). It
is unknown what aspect of higher SES is relevant but
higher age of exposure to infectious agents has been
hypothesized.
16,22,36,37
Ionizing radiation (in utero) In past studies, there was a consistent, increased risk
(about 1.5 fold) of leukemia associated with prenatal
diagnostic x-ray exposure. However, this is unlikely to be
an important risk factor for childhood leukemia today due
to fewer x-rays, increased shielding, and lower radiation
levels.
16,22,36,38,39
Ionizing radiation postnatal
(therapeutic)
Therapeutic radiation for such conditions as tinea capitis
and thymus enlargement has been associated with an
increased risk.
16,22,36,40
Down syndrome,
neurofibromatosis, Shwachman
syndrome, Bloom syndrome,
ataxia telangiectasia,
Langerhans cell histiocytosis, and
Klinefelter syndrome
Increased occurrence is associated with these genetic
conditions and is particularly apparent in children with
Down syndrome for whom there is a reported 20-fold
increased risk of leukemia.
16,22,26,36
High birth weight (> 4000 grams) Several studies have reported an elevated risk
(approximately 2-fold) in larger babies, particularly for
children diagnosed younger than two years of age.
16,22,36,41
Maternal history of fetal loss
prior to the birth of the index
child
Approximately 2-5 fold increased risk of leukemia has
been noted in a few studies; particularly in children
diagnosed younger than two years of age.
16,18,22,36,42
Maternal age > 35 at pregnancy A slight increased risk has been somewhat inconsistently
associated with older maternal age.
16,22,36
First born or only child Slight increased risk reported but birth order may be
surrogate marker for exposure to infectious agent.
16,22,36
Known risk factors
Factors for which evidence is
suggestive but not conclusive
ICCC I
30
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
Table I.5 (cont’d): Current knowledge on causes of acute lymphoblastic leukemia (ALL)
Smoking prior to and during
pregnancy
Some studies have reported an increased risk associated
with maternal smoking during pregnancy but others have
not. A few recent studies have suggested a modest
increased risk (about 1.5 fold) associated with paternal
smoking prior to pregnancy.
16,22,36,43-48
Parental occupations and
occupational exposures
Isolated reports associated with parental exposure to
motor vehicle exhaust, hydrocarbons, and paints. This is
the subject of several current epidemiologic studies.
16,22,36
Postnatal infections Evidence is very inconsistent. 16,22
Diet A few reports have suggested that meat consumption
(particularly, cured meats) is associated with an increased
risk. Maternal diet and childhood leukemia has not been
explored in any detail and further study is warranted.
49,50
Electromagnetic fields A few studies have reported a slight increased risk for
children living near high voltage power lines; others have
reported no association. A recent large study of U.S.
children with ALL found little or no association between
risk of ALL and electromagnetic field exposure. Other
large epidemiologic studies evaluating this exposure are
ongoing.
16,22,51-54
Vitamin K prophylaxis in
newborns
Although an increased risk of ALL was first reported in
the early 1990's, several large studies since then have
found no association.
16,22,55-60
Maternal alcohol consumption
during pregnancy
Unlikely to be an important risk factor for ALL (but see
AML).
16,22
Postnatal use of chloramphenicol One study reported quite substantial increased risks
(approximately 10-fold) with postnatal use of this broad-
spectrum antibiotic.
61
Ultrasound 16,22
*Note that the majority of these risk factors have been reviewed recently in references 16,22,36; only
selected references are presented for additional reading.
Factors for which evidence is
inconsistent or limited
Factors unrelated to risk
Different risk factors are emerging for
childhood AML that distinguish the disease
from ALL, and this may provide avenues
for future epidemiological studies. For
example, exposure to specific chemotherapy
agents has been associated with an in-
creased risk of childhood AML, in contrast
to the rarity of treatment-related ALL [22-
24]. With this information, it may be
possible to design epidemiological studies to
examine exposures to environmental
agents that have a biologic nature that is
similar to these chemotherapy agents [14].
Further
,
associations with factors such as
benzene and pesticides that have emerged
in a few studies suggest that childhood
AML may share risk factors with adult
AML, and this is being investigated in
several large epidemiological studies [25].
Finally, as for ALL, the associations of AML
with genetic syndromes are compelling, as
illustrated by the magnitude of risk of AML
in Down syndrome [26].
SUMMARY
ALL is by far the most common type of
leukemia occurring in children and shows a
distinctive age-distribution pattern, with a
marked incidence peak at 2-3 years of age.

ICCC ILEUKEMIA
31
National Cancer Institute SEER Pediatric Monograph
Known risk factors
Factors for which evidence is
suggestive but not conclusive
Factors for which evidence is
inconsistent or limited
Table I.6: Current knowledge on causes of acute myeloid leukemia (AML)
Exposure or Characteristic Comments References
Race The highest incidence rates are reported in the
Hispanic children.
16,22,36
Chemotherapeutic agents Increased risk is associated with prior exposure to
alkylating agents or epipodophyllotoxins.
16,22,36,62,63
Ionizing radiation (
in utero
) In past studies, there was a consistent, increased risk
(about 1.5 fold) of leukemia associated with prenatal
diagnostic x-ray exposure. However, this is unlikely
to be an important risk factor for childhood leukemia
today due to fewer x-rays, increased shielding, and
lower radiation levels.
16,22,36
Down syndrome, neurofibromatosis,
Shwachman syndrome, Bloom
syndrome, familial monosomy 7,
Kostmann granulocytopenia, Fanconi
anemia
Increased occurrence associated with these genetic
conditions, particularly with Down syndrome. One
report suggests as high as a 500-fold increased risk of
a specific type of AML in Down syndrome.
16,22,26,36
Maternal alcohol consumption during
pregnancy
Three studies have reported an increased risk
(approximately 1.5-2 fold) in mothers who drank
alcoholic beverages during pregnancy. These
associations have been particularly apparent in
children diagnosed younger than three years of age.
16,22,36,64
Parental and child exposure to
pesticides
Increased risk has been noted in a few studies and in
adult AML data; subject of several current
investigations.
16,22,25,36,45
Parental exposure to benzene Exposure has been associated with an increased risk
in several studies; again follows adult AML data; also
subject of several current investigations.
16,22,36,45
Maternal use of recreational drugs
during pregnancy
One report suggested that maternal marijuana use
during pregnancy was associated with increased risk.
65
Radon A few correlational studies have suggested an
increased risk of childhood and adult AML in areas
with high radon concentrations; this is a subject of
several current epidemiologic studies of AML.
16,22,36
Postnatal use of chloramphenicol As in ALL, one study found quite substantial
increased risks of AML (approximately 10-fold) with
postnatal use of this broad-spectrum antibiotic.
61
*Note that the majority of these risk factors have been reviewed recently in references [16,22,36]; only
elected references are presented for additional reading.

ICCC I
32
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
The peak at 2-3 years of age is much less
apparent for black children than for white
children, with this difference accounting for
the substantially lower incidence of ALL
observed for black children. By contrast
with ALL, the age-distribution pattern for
AML shows highest rates in the first two
years of life, with decreasing incidence until
10 years of age followed by increasing rates
thereafter. Again in contrast to ALL, the
incidence of AML in black children and
white children is similar.
The improvement in survival for chil-
dren with ALL over the past 35 years is one
of the great success stories of clinical oncol-
ogy. Survival rates for childhood ALL were
below 5% in the early 1960s, but are now
approaching 80% [35]. Outcome for chil-
dren with AML has also improved, but 5-
year survival rates have increased to only
the 40% range. Black children with ALL
have poorer outcome than do white chil-
dren, but for AML there is similar outcome
for black children and white children. Since
the peak in ALL incidence at 2-3 years of
age is much lower for black children than
for white children and since these ALL
cases in young children are known to have
the most favorable prognosis, the poorer
outcome for black children may reflect in
part a different distribution of biological
subtypes of ALL in black children compared
to white children.
Reference List
1. Margolin J, Poplack D: Acute lymphoblastic
leukemia. In Principles and Practice of Pediat-
ric Oncology (Pizzo P, Poplack D, eds). Philadel-
phia: Lippincott-Raven, 1997, pp 409-462.
2. Golub T, Weinstein H, Grier H: Acute myelog-
enous leukemia. In Principles and Practice of
Pediatric Oncology (Pizzo P, Poplack D, eds).
Philadelphia: Lippincott-Raven, 1997, pp 463-
482.
3. Cheson BD, Cassileth PA, Head DR, et al:
Report of the National Cancer Institute-
sponsored workshop on definitions of diagnosis
and response in acute myeloid leukemia. J
Clin Oncol 8:813-9, 1990.
4. Smith M, Chen T, Simon R: Age-specific
incidence of acute lymphoblastic leukemia in
U.S. children: in utero initiation model. J Natl
Cancer Inst 89:1542-1544, 1997.
5. Gale KB, Ford AM, Repp R, et al: Backtracking
leukemia to birth: identification of clonotypic
gene fusion sequences in neonatal blood spots.
Proc Natl Acad Sci U S A 94:13950-4, 1997.
6. Ford AM, Bennett CA, Price CM, et al: Fetal
origins of the TEL-AML1 fusion gene in
identical twins with leukemia. Proc Natl Acad
Sci U S A 95:4584-8, 1998.
7. Ries L, Kosary C, Hankey B, et al: SEER
Cancer Statistics Review, 1973-1994. Bethesda,
MD: National Cancer Institute, 1997.
8. Hess JL, Zutter MM, Castleberry RP, et al:
Juvenile chronic myelogenous leukemia. Am J
Clin Pathol 105:238-48, 1996.
9. Arico M, Biondi A, Pui CH: Juvenile
myelomonocytic leukemia [see comments].
Blood 90:479-88, 1997.
10. Gurney JG, Davis S, Severson RK, et al: Trends
in cancer incidence among children in the U.S.
Cancer 78:532-41, 1996.
11. Bunin GR, Feuer EJ, Witman PA, et al: Increas-
ing incidence of childhood cancer: report of 20
years experience from the greater Delaware
Valley Pediatric Tumor Registry. Paediatr
Perinat Epidemiol 10:319-38, 1996.
12. Linet MS, Devesa SS: Descriptive epidemiology
of childhood leukaemia. Br J Cancer 63:424-9,
1991.
13. Chow W-H, Linet M, Liff J, et al: Cancers in
children. In Cancer Epidemiology and Preven-
tion (Schottenfeld D, Frameni Jr. J, eds). New
York: Oxford, 1996, pp 1331-69.
14. Ross J, Potter J, Robison L: Infant leukemia,
topoisomerase II inhibitors, and the MLL gene.
J Natl Cancer Inst 86:1678-1680, 1994.
15. Ross JA, Potter JD, Reaman GH, et al: Mater-
nal exposure to potential inhibitors of DNA
topoisomerase II and infant leukemia (United
States): a report from the Children’s Cancer
Group. Cancer Causes Control 7:581-90, 1996.
16. Ross JA, Davies SM, Potter JD, et al: Epidemi-
ology of childhood leukemia, with a focus on
infants. Epidemiol Rev 16:243-72, 1994.
17. Ross JA, Potter JD, Shu XO, et al: Evaluating
the relationships among maternal reproductive
history, birth characteristics, and infant
leukemia: a report from the Children’s Cancer
Group. Ann Epidemiol 7:172-9, 1997.
18. Yeazel MW, Buckley JD, Woods WG, et al:
History of maternal fetal loss and increased
risk of childhood acute leukemia at an early
age. A report from the Childrens Cancer
Group. Cancer 75:1718-27, 1995.

ICCC ILEUKEMIA
33
National Cancer Institute SEER Pediatric Monograph
19. Yeazel MW, Ross JA, Buckley JD, et al: High
birth weight and risk of specific childhood
cancers: a report from the Children’s Cancer
Group. J Pediatr 131:671-7, 1997.
20. Greaves M: Aetiology of acute leukemia. Lancet
349:344-349, 1997.
21. Smith MA, Simon R, Strickler HD, et al:
Evidence that childhood acute lymphoblastic
leukemia is associated with an infectious agent
linked to hygiene conditions. Cancer Causes
Control 9:285-98, 1998.
22. Robison L, Ross J: Epidemiology of leukaemias
and lymphomas in childhood. In Bailliere’s
Clinical Paediatrics (Chessells J, Hann I, eds).
London: WB Saunders Co., 1995, pp pp. 639-
657.
23. Smith M, Rubinstein L, Ungerleider R:
Therapy-related acute myeloid leukemia
following treatment with epipodophyllotoxins:
Estimating the risks. Med Pediatr Oncol 23:86-
98, 1994.
24. Hunger S, Sklar J, Link M: Acute lymphoblas-
tic leukemia occurring as a second malignant
neoplasm in childhood: Report of three cases
and review of the literature. J Clin Oncol
10:156-163, 1992.
25. Buckley JD, Robison LL, Swotinsky R, et al:
Occupational exposures of parents of children
with acute nonlymphocytic leukemia: a report
from the Childrens Cancer Study Group.
Cancer Res 49:4030-7, 1989.
26. Zipursky A, Brown E, Christensen H, et al:
Leukemia and/or myeloproliferative syndrome
in neonates with Down syndrome. Semin
Perinatol 21:97-101, 1997.
27. Tubergen D, Gilchrist G, O’Brien R, et al:
Improved outcome with delayed intensification
for children with acute lymphoblastic leukemia
and intermediate presenting features: A
Childrens Cancer Group Phase III Trial. J Clin
Oncol 11:527-537, 1993.
28. Land VJ, Shuster JJ, Crist WM, et al: Compari-
son of two schedules of intermediate-dose
methotrexate and cytarabine consolidation
therapy for childhood B-precursor cell acute
lymphoblastic leukemia: a Pediatric Oncology
Group study. J Clin Oncol 12:1939-45, 1994.
29. Mahoney DH, Jr., Shuster J, Nitschke R, et al:
Intermediate-dose intravenous methotrexate
with intravenous mercaptopurine is superior to
repetitive low-dose oral methotrexate with
intravenous mercaptopurine for children with
lower-risk B-lineage acute lymphoblastic
leukemia: a Pediatric Oncology Group phase
III trial. J Clin Oncol 16:246-54, 1998.
30. Trueworthy R, Shuster J, Look T, et al: Ploidy
of lymphoblasts is the strongest predictor of
treatment outcome in B-progenitor cell acute
lymphoblastic leukemia of childhood: A Pediat-
ric Oncology Group Study. J Clin Oncol 10:606-
613, 1992.
31. Rubnitz JE, Shuster JJ, Land VJ, et al: Case-
control study suggests a favorable impact of
TEL rearrangement in patients with B-lineage
acute lymphoblastic leukemia treated with
antimetabolite-based therapy: a Pediatric
Oncology Group study. Blood 89:1143-6, 1997.
32. Rubnitz JE, Downing JR, Pui CH, et al: TEL
gene rearrangement in acute lymphoblastic
leukemia: a new genetic marker with prognos-
tic significance. J Clin Oncol 15:1150-7, 1997.
33. Rubnitz JE, Link MP, Shuster JJ, et al: Fre-
quency and prognostic significance of HRX
rearrangements in infant acute lymphoblastic
leukemia: a Pediatric Oncology Group study.
Blood 84:570-3, 1994.
34. Hilden JM, Frestedt JL, Moore RO, et al:
Molecular analysis of infant acute lymphoblas-
tic leukemia: MLL gene rearrangement and
reverse transcriptase-polymerase chain
reaction for t(4; 11)(q21; q23). Blood 86:3876-82,
1995.
35. Harras A: Cancer: Rates and Risks. In National
Institutes of Health publication No. 96-691.
Bethesda, MD: National Cancer Institute, 1996.
36. Sandler DP, Ross JA: Epidemiology of acute
leukemia in children and adults. Semin Oncol
24:3-16, 1997.
37. McWhirter W: The relationship of incidence of
incidence of childhood lymphoblastic leukaemia
to social class. Br J Cancer 46:640-645, 1982.
38. MacMahon B, Newill V: Birth characteristics of
children dying of malignant neoplasms. J Natl
Cancer Inst 37:687-698, 1962.
39. Ford D, Paterson J, Treuting W: Fetal exposure
to diagnostic x-rays, and leukemia and other
malignant disease of childhood. J Natl Cancer
Inst 22:1093-1104, 1959.
40. Ron E, Modan B: Thyroid and other neoplasms
following childhood scalp irradiation. In
Radiation Carcinogenesis, Epidemiology and
Biological Significance (Boice J, Fraumeni J,
eds). New York, NY: Raven Press, 1984, pp 139.
41. Ross JA, Perentesis JP, Robison LL, et al: Big
babies and infant leukemia: a role for insulin-
like growth factor-1? Cancer Causes Control
7:553-9, 1996.
42. Kaye SA, Robison LL, Smithson WA, et al:
Maternal reproductive history and birth
characteristics in childhood acute lymphoblas-
tic leukemia. Cancer 68:1351-5, 1991.
43. Neutel CI, Buck C: Effect of smoking during
pregnancy on the risk of cancer in children. J
Natl Cancer Inst 47:59-63, 1971.
44. van Steensel-Moll HA, Valkenburg HA,
Vandenbroucke JP, et al: Are maternal fertility
problems related to childhood leukaemia? Int J
Epidemiol 14:555-9, 1985.

ICCC I
34
National Cancer Institute
SEER Pediatric Monograph
LEUKEMIA
45. Shu XO, Gao YT, Brinton LA, et al: A popula-
tion-based case-control study of childhood
leukemia in Shanghai. Cancer 62:635-44, 1988.
46. Ji BT, Shu XO, Linet MS, et al: Paternal
cigarette smoking and the risk of childhood
cancer among offspring of nonsmoking mothers.
J Natl Cancer Inst 89:238-44, 1997.
47. Sorahan T, Prior P, Lancashire RJ, et al:
Childhood cancer and parental use of tobacco:
deaths from 1971 to 1976. Br J Cancer 76:1525-
31, 1997.
48. Sorahan T, Lancashire RJ, Hulten MA, et al:
Childhood cancer and parental use of tobacco:
deaths from 1953 to 1955. Br J Cancer 75:134-
8, 1997.
49. Peters JM, Preston-Martin S, London SJ, et al:
Processed meats and risk of childhood leuke-
mia (California, USA). Cancer Causes Control
5:195-202, 1994.
50. Sarasua S, Savitz DA: Cured and broiled meat
consumption in relation to childhood cancer:
Denver, Colorado (United States) [see com-
ments]. Cancer Causes Control 5:141-8, 1994.
51. Wertheimer N, Leeper E: Electrical wiring
configurations and childhood cancer. Am J
Epidemiol 109:273-84, 1979.
52. Savitz DA, Wachtel H, Barnes FA, et al: Case-
control study of childhood cancer and exposure
to 60-Hz magnetic fields [see comments]. Am J
Epidemiol 128:21-38, 1988.
53. Verkasalo PK, Pukkala E, Hongisto MY, et al:
Risk of cancer in Finnish children living close
to power lines [see comments]. BMJ 307:895-9,
1993.
54. Linet MS, Hatch EE, Kleinerman RA, et al:
Residential exposure to magnetic fields and
acute lymphoblastic leukemia in children [see
comments]. N Engl J Med 337:1-7, 1997.
55. Golding J, Greenwood R, Birmingham K, et al:
Childhood cancer, intramuscular vitamin K,
and pethidine given during labour [see com-
ments]. BMJ 305:341-6, 1992.
56. Klebanoff MA, Read JS, Mills JL, et al: The
risk of childhood cancer after neonatal expo-
sure to vitamin K [see comments]. N Engl J
Med 329:905-8, 1993.
57. Ekelund H, Finnstrom O, Gunnarskog J, et al:
Administration of vitamin K to newborn
infants and childhood cancer. BMJ 307:89-91,
1993.
58. McKinney PA, Juszczak E, Findlay E, et al:
Case-control study of childhood leukaemia and
cancer in Scotland: findings for neonatal
intramuscular vitamin K [see comments]. BMJ
316:173-7, 1998.
59. Parker L, Cole M, Craft AW, et al: Neonatal
vitamin K administration and childhood cancer
in the north of England: retrospective case-
control study [see comments]. BMJ 316:189-93,
1998.
60. Passmore SJ, Draper G, Brownbill P, et al:
Case-control studies of relation between
childhood cancer and neonatal vitamin K
administration [see comments]. BMJ
316:178-84, 1998.
61. Shu XO, Gao YT, Linet MS, et al: Chloram-
phenicol use and childhood leukaemia in
Shanghai. Lancet 2:934-7, 1987.
62. Pui CH, Behm FG, Raimondi SC, et al:
Secondary acute myeloid leukemia in chil-
dren treated for acute lymphoid leukemia
[see comments]. N Engl J Med 321:136-42,
1989.
63. Smith M, McCaffrey R, Karp J: The secondary
leukemias: Challenges and research direc-
tions. J Natl Cancer Inst 88:407-418, 1996.
64. Shu XO, Ross JA, Pendergrass TW, et al:
Parental alcohol consumption, cigarette
smoking, and risk of infant leukemia: a
Childrens Cancer Group study. J Natl Cancer
Inst 88:24-31, 1996.
65. Robison LL, Buckley JD, Daigle AE, et al:
Maternal drug use and risk of childhood
nonlymphoblastic leukemia among offspring.
An epidemiologic investigation implicating
marijuana (a report from the Childrens
Cancer Study Group). Cancer 63:1904-11,
1989.

ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
35
National Cancer Institute
SEER Pediatric Monograph
Constance L. Percy, Malcolm A. Smith, Martha Linet, Lynn A. Gloeckler Ries, Debra L. Friedman
HIGHLIGHTS
Incidence
♦ Approximately 15% of childhood malignancies were lymphomas making them the
third most frequent type of cancer in children following leukemia and malignant
brain tumors. The percentage of childhood cancer that were lymphoma varied by
age from only 3% for children younger than 5 years of age to 24% for 15-19 year olds
(Figure II.1).
♦ The two predominant types of lymphomas were Hodgkin’s disease and the non-
Hodgkin’s lymphomas (NHL). For younger children NHL was more frequent than
Hodgkin’s disease, while the reverse was true for adolescents (Figure II.3).
♦ In the US, approximately 1,700 children and adolescents younger than 20 years of
age are diagnosed with lymphomas each year of which approximately 850-900 are
cases of Hodgkin’s disease and 750-800 are cases of NHL.
♦ The most common subtypes of Hodgkin’s disease were nodular sclerosis (70% of
cases); mixed cellularity (16% of cases); lymphocytic predominance (7% of cases);
cases not otherwise specified (NOS) (6% of cases); and lymphocytic depletion subtype
(<2% of cases) among children younger than 20 years of age. The relative frequen-
cies of the mixed cellularity and nodular sclerosis subtypes were age and sex depen-
dent (Figure II.2 and Table II.2).
♦ The incidence of Hodgkin’s disease for children and adolescents younger than 20
years of age decreased slightly between 1975 and 1995, from 14.5 per million (1975-
79) to 12.1 per million (1990-95) (Table II.3).
♦ The non-Hodgkin’s lymphomas of children are a heterogeneous group of tumors,
with Burkitt’s and Burkitt-like tumors predominating among 5-14 year olds, and
with diffuse large cell lymphomas being the most common subtype among 15-19
year olds (Figure II.9).
♦ The incidence of NHL varied much less by age than Hodgkin’s disease (Figure II.3).
NHL incidence increased up until age 4 years where it reached a plateau of approxi-
mately 10 per million (Figure II.4), which was maintained until the second decade of
life when rates increased again.
♦ The incidence of NHL was higher in males than females (Figure II.10) and higher
among whites than blacks (Figure II.12).
♦ The incidence of NHL among children younger than 15 years of age was fairly
constant over the past 21 years, while there appeared to have been a slight increase
in incidence for the 15-19 year old population (Table II.3 and Figure II.13).
Survival
♦ The 5-year survival rate was 91% for Hodgkin’s disease (Figure II.8) for children
and adolescents younger than 20 years of age, compared to 72% for NHL (Figure
II.14).
♦ The 5-year survival rate for those younger than 20 years of age with NHL increased
from 56% in 1975-84 to 72% in 1985-94 (Figure II.14).
Risk factors
♦ For Hodgkin’s disease arising in young adults, genetic susceptibility may be a factor
for some cases, based on the greatly increased risk for Hodgkin’s disease in young
adult monozygotic twins of patients with Hodgkin’s disease compared to the risk of
dizygotic twins of patients with Hodgkin’s disease (Table II.4).
♦ Congenital immunodeficiency syndromes and acquired immunodeficiency syndrome
(AIDS) are associated with an increased risk of NHL (Table II.6).
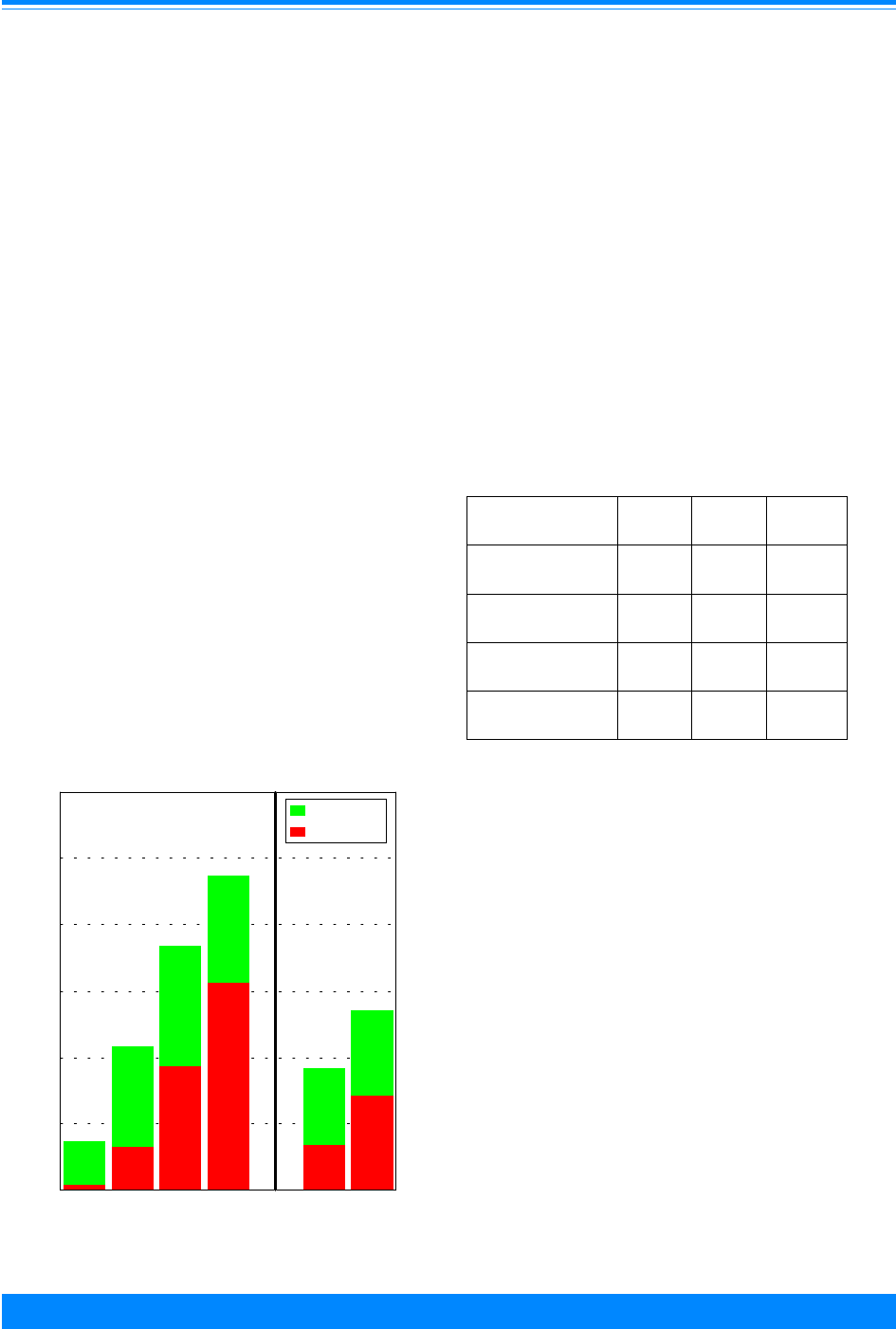
36
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
INTRODUCTION
The lymphomas, combining Hodgkin’s
disease and the non-Hodgkin’s lymphomas
(NHL), are the third most frequent type of
cancer in children following leukemia and
malignant brain tumors. NHL and
Hodgkin’s disease are both malignancies of
lymphoid cells, and each includes distinc-
tive biological subtypes. In the US, ap-
proximately 1,700 children and adolescents
younger than 20 years of age are diagnosed
with lymphomas each year, of which 850-
900 are Hodgkin’s disease and 750-800 are
non-Hodgkin’s lymphoma. The lymphomas
account for 10% of malignancies among
children younger than 15 years of age and
15% of malignancies among those younger
than 20 years of age. Figure II.1 illustrates
that the contribution of Hodgkin’s disease
and the non-Hodgkin’s lymphomas to the
overall childhood cancer burden is mark-
edly age dependent, increasing from only
3% of cancers among children younger than
5 years of age to 24% of cancers arising
among those 15-19 years of age.
MATERIAL AND METHODS
The International Classification for
Childhood Cancers (ICCC) Group II of
Lymphomas and Reticuloendothelial Neo-
plasms is divided into 5 subgroups [1]:
a. Hodgkin’s disease;
b. Non-Hodgkin’s lymphoma;
c. Burkitt’s lymphoma;
d. Miscellaneous lymphoreticular
neoplasms; and
e. Unspecified lymphomas.
Since only 3% of the registered child-
hood lymphomas and reticuloendothelial
neoplasms in the Surveillance, Epidemiol-
ogy and End Results (SEER) program for
the period 1975-95 were not histologically
confirmed, all cases were included in the
analyses. Table II.1 shows the frequency
distribution of these cases by sex among
children younger than 20 years of age
diagnosed with lymphoma. For the pur-
poses of this chapter, Hodgkin’s disease
(defined by subgroup IIa) and NHL (defined
by subgroups IIb, IIc, and IIe) are consid-
ered separately because of their distinctive
nature. The miscellaneous lymphoreticular
neoplasms (IId) are excluded from further
analysis as there were few cases (only 79
cases) in this subgroup from 1975-95.
Table II.1: Number of cases of lymphoma by type
and sex, age <20, all races, SEER, 1975-95
Total Males
(%)
Females
(%)
Total 4595 2734
(59%)
1861
(41%)
Hodgkin’s (IIa) 2613 1353
(52%)
1260
(48%)
NHL (IIb,c,e) 1903 1334
(70%)
569
(30%)
Miscellaneous (IId) 79 47
(59%)
32
(41%)
Figure II.1: Hodgkin's disease and NHL as a percent of
total childhood cancer, by age, all races, both sexes
SEER, 1990-95
0
3
9
16
3
7
3
8
9
8
6
6
<5 5-9 10-14 15-19 <15 <20
Age (in years) at diagnosis
0
5
10
15
20
25
30
Relative percent
Hodgkin's
NHL
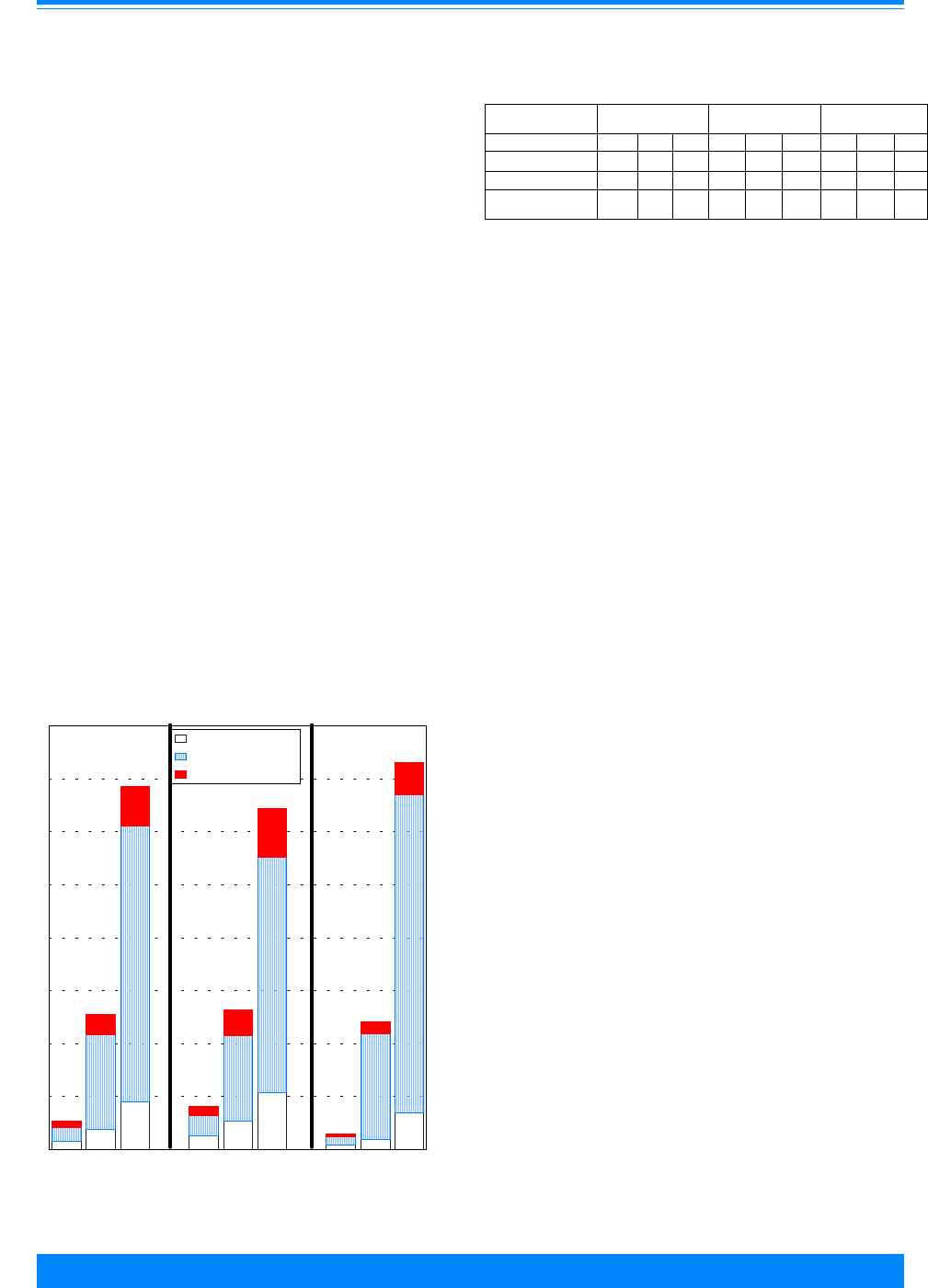
37
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
Hodgkin’s disease is an unusual malig-
nancy of lymphoid cells whose distinctive
hallmark is the Reed-Sternberg cell. In
most cases Reed-Sternberg cells appear to
be clonal in nature and from the B lympho-
cyte lineage, as evidenced by immunoglobu-
lin gene rearrangements in these cells [2-5].
Reed-Sternberg cells account for only a
small percentage of the tumor mass [6],
with most of the tumor composed of a
reactive infiltrate of lymphocytes, plasma
cells, and eosinophils. The Reed-Sternberg
cells of some patients with Hodgkin’s
disease contain copies of the Epstein-Barr
virus (EBV) genome [7,8]. Detection of
EBV in Reed-Sternberg cells is more com-
mon among cases diagnosed in young
children and for cases of the mixed cellular-
ity subtype [7,8] (see Histologic subtype
and Risk Factors sections below).
Histologic subtype
Childhood Hodgkin’s disease, similar to
that arising in adults, is usually classified
according to the Rye classification scheme,
which includes four histologic subtypes:
lymphocytic predominance, mixed cellular-
ity, lymphocytic depletion, and nodular
sclerosis [9]. The lymphocytic predomi-
nance subtype is now recognized as having
a distinctive biological and clinical behavior
from the other subtypes of Hodgkin’s dis-
ease and is relatively uncommon among
children [10,11]. Among children younger
than 20 years of age diagnosed with
Hodgkin’s disease during 1975-95, the
nodular sclerosis subtype was by far the
most common and accounted for 70% of all
cases. Mixed cellularity was the second
most common subtype (16% of cases),
followed by lymphocytic predominance (7%
of cases), and cases not otherwise specified
(NOS), (6% of cases). The lymphocytic
depletion subtype was distinctly uncommon
(<2% of cases) among Hodgkin’s disease
cases diagnosed in those younger than 20
years of age.
The relative frequencies of the mixed
cellularity and nodular sclerosis subtypes
were age and sex dependent. Mixed cellu-
larity subtype was more common among
children younger than 10 years of age (32%
of all Hodgkin’s cases) than among those
diagnosed at 10-14 or 15-19 years of age
(15% and 13% of Hodgkin’s cases, respec-
tively) (Table II.2 and Figure II.2). Mixed
cellularity was also more common among
males than females in each age group.
Table II.2: Percent distribution of Hodgkin’s by subtype, age and
sex all races, SEER, 1975-95
Age (in years) at
diagnosis
<10 10-14 15-19
Both M F Both M F Both M F
Mixed Cellularity
32% 34% 25% 15% 21% 9% 13% 17% 10%
Nodular Sclerosis
46% 44% 51% 69% 59% 79% 74% 67% 81%
Lymphocytic
Predominance
14% 15% 12% 7% 10% 3% 5% 7% 2%
Figure II.2: Hodgkin's disease age-adjusted* incidence
rates by Rye classification, sex and age
all races, SEER (10 areas), 1977-95
0.8
1.9
4.5
1.3
2.7
5.4
0.4
1
3.5
1.3
8.9
26
1.9
8
22.2
0.8
9.9
30
0.6
1.9
3.8
0.9
2.5
4.6
0.3
1.2
3
<10 10-14 15-19 <10 10-14 15-19 <10 10-14 15-19
0
5
10
15
20
25
30
35
40
Average annual rate per million
Mixed cellularity
Nodular sclerosis
Other
Both Sexes
Male
Female
*Adjusted to the 1970 US standard population
HODGKIN’S DISEASE

38
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
Nodular sclerosis subtype accounted for the
vast majority (81%) of Hodgkin’s disease
among 15-19 year old females, and a some-
what lower percentage (approximately
67%) of Hodgkin’s disease among 15-19
year old males (Table II.2).
Age-specific incidence
The overall annual incidence rate for
Hodgkin’s disease among children younger
than 20 years of age was 12.1 per million
for the years 1990-95. For Hodgkin’s
disease, the contribution to overall child-
hood cancer incidence was less than 1%
among children younger than 5 years of
age, but increased to 16% for 15-19 year
olds (Figure II.1). The incidence of
Hodgkin’s disease was markedly age depen-
dent (Figures II.3 and II.4), with incidence
increasing from less than one per million
for children in the first 3 years of life, to
Figure II.3: Hodgkin's disease and NHL age-specific and age-adjusted*
incidence rates by age, all races, both sexes
SEER, 1990-95
6.4
8.2
10.7
16.3
8.6
10.5
0.9
3.6
11.2
32
5.5
12.1
<5 5-9 10-14 15-19 <15 <20
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
Average annual rate per million
NHL
Hodgkin's
*Adjusted to the 1970 US standard population
Figure II.4: Hodgkin's disease and NHL age-specific
incidence rates, all races, both sexes
SEER, 1976-84 and 1986-94
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
10
20
30
40
50
Average annual rate per million
NHL (II b,c,e)
Hodgkin's
,
(

39
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
43.2 per million for 19 year olds (Figure
II.4).
1
While the overall incidence of
Hodgkin’s disease for children younger than
20 years of age was similar to that for NHL
(12.1 versus 10.5 per million, respectively)
(Figure II.3), the incidence of Hodgkin’s
disease was 2-3 fold higher than NHL
among 15-19 year olds, while the incidence
of NHL was higher among those younger
than 10 years of age (Figure II.4).
Sex-specific incidence
The incidence of Hodgkin’s disease was
slightly higher among females than males
for children younger than 20 years of age
during 1990-95 (M/F = 0.9) (Figure II.5).
However, the differential by sex was age
dependent. Among children younger than
15 years of age, Hodgkin’s disease was
more common among males (M/F = 1.3),
with the differential greatest for children
younger than 5 years of age (M/F = 5.3).
However, for children diagnosed with
Hodgkin’s disease at ages 15-19 years,
Hodgkin’s disease incidence was higher
among females (M/F = 0.8).
Black-white differences in incidence
When considering children younger
than 20 years of age, black children had a
lower incidence of Hodgkin’s disease than
white children (Figure II.6). However, the
incidence was very similar for black and
white children younger than 10 years of
age. For those over 10 years of age, the
ratio of white to black incidence was ap-
proximately 1.4:1.
Figure II.5: Hodgkin's disease age-specific
and age-adjusted* incidence rates by age
and sex with male to female ratios (M/F)
all races, SEER, 1990-95
1.6
4.2
11.9
28.3
6.2
11.6
0.3
3
10.5
36
4.9
12.6
<5 5-9 10-14 15-19 <15 < 20
Age (in years) at diagnosis
0
10
20
30
40
Average annual rate per million
Male
Female
M/F=5.3
M/F=1.4
M/F=1.1
M/F=0.8
M/F=1.3
M/F=0.9
*Adjusted to the 1970 US standard population
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.
W/B=1.3
Figure II.6: Hodgkin's disease age-specific and
age-adjusted* incidence rates by age with white
to black ratios (W/B) both sexes, SEER, 1990-95
1
4.1
13
36.5
6.4
13.8
0
4.5
9.5
26.9
5
10.4
<5 5-9 10-14 15-19 <15 <20
Age (in years) at diagnosis
0
10
20
30
40
Average annual rate per million
White
Black
W/B=0.9
W/B=1.4
W/B=1.4
W/B=1.3
*Adjusted to the 1970 US standard population
W/B=1.3
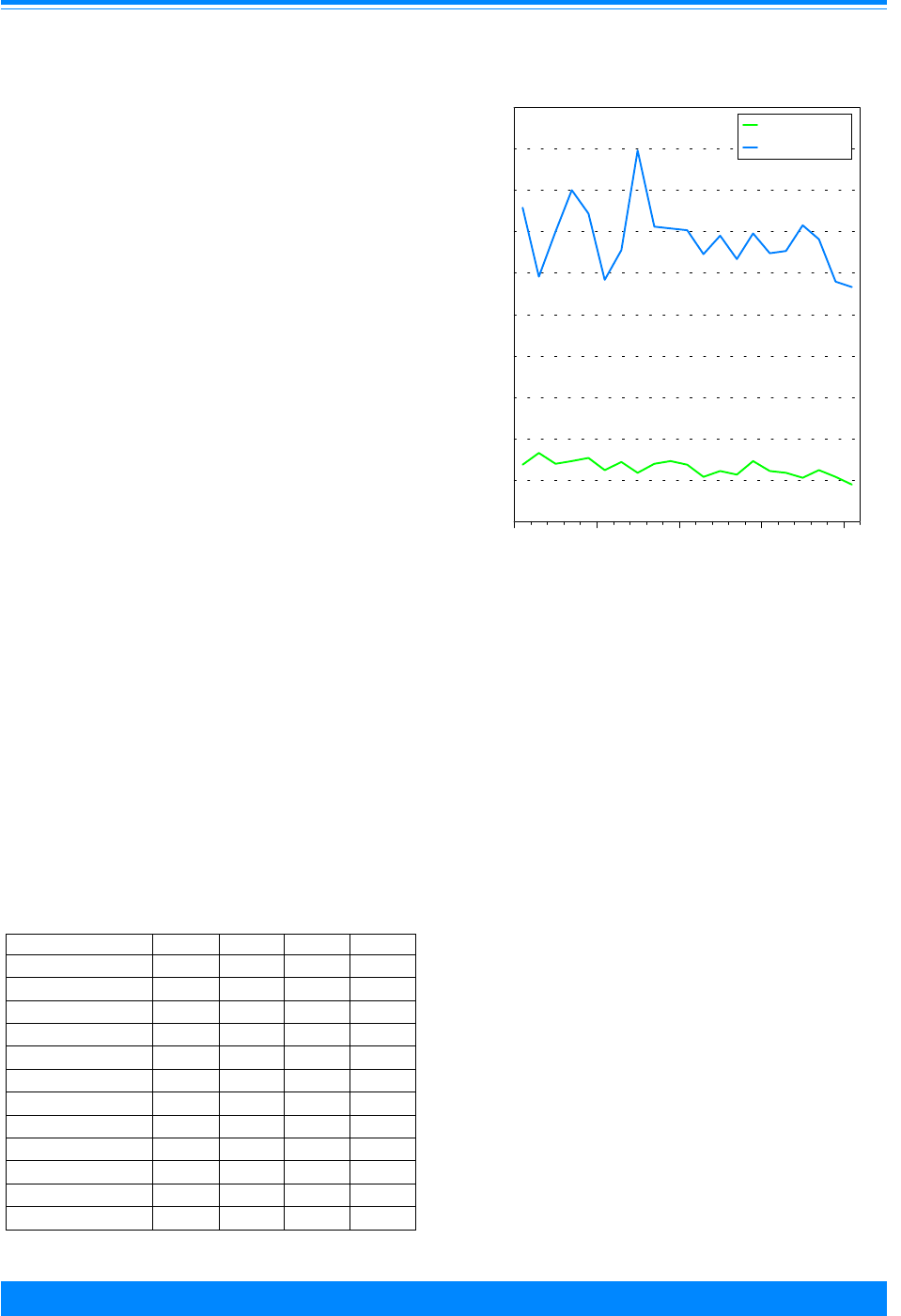
40
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
Table II.3: Hodgkin’s and NHL age-adjusted
*
incidence rates
all races, both sexes, SEER, 1975-79 through 1990-95
1975-79 1980-84 1985-89 1990-95
Hodgkin’s <15 years
7.4 6.7 6.3 5.5
Males
8.4 8.0 7.5 6.2
Females
6.4 5.3 5.1 4.9
Hodgkin’s <20 years
14.5 13.8 13.1 12.1
Males
14.8 14.8 13.4 11.6
Females
14.2 12.8 12.8 12.6
NHL <15 years
8.5 8.1 8.6 8.5
Males
11.7 12.6 11.7 12.7
Females
5.2 3.4 5.3 4.2
NHL <20 years
9.1 9.7 10.0 10.5
Males
12.1 14.0 13.4 14.6
Females
5.9 5.2 6.5 6.1
*
Adjusted to the 1970 US standard population
TRENDS
The age-adjusted incidence rate for
Hodgkin’s disease for children younger than
20 years of age decreased from 14.5 per
million in 1975-79 to 12.1 per million in
1990-95 (Table II.3). The incidence for
children younger than 15 years of age
declined during this period from approxi-
mately 7.4 per million to 5.5 per million.
The incidence for 15-19 year olds declined
from 35.9 per million (1975-79) to 32.0 per
million (1990-95) (Figure II.7). The decline
in Hodgkin’s incidence rates for the
younger than 15 year age group occurred
for both males and females [estimated
annual percentage change (EAPC) of -2.0%
and -1.4%, respectively]. The decline in
incidence for 15-19 year olds appeared to be
greater for males (EAPC = -1.4%) than for
females (EAPC = -0.1%). A similar pattern
of declining rates for males and stable rates
for females was observed in regions of the
United Kingdom for the years 1984-93 [12].
Another report described little change or
slight decline in the occurrence of Hodgkin’s
disease among 20 populations from around
the world for the period from the early
1970s to the 1980s [13]. In contrast to
these reports, increasing incidence rates for
the 15-24 year olds were reported for males
and females in four geographical areas of
the US for the period from 1947-50 to 1984-
88 [13].
Figure II.7: Trends in Hodgkin's disease age-adjusted*
incidence rates by age, all races, both sexes, SEER, 1975-95
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
1975 1980 1985 1990 1995
Year of diagnosis
0
5
10
15
20
25
30
35
40
45
50
Average annual rate per million
<15 Years
15-19 Years
(
&
*Adjusted to the 1970 US standard population
SURVIVAL
For cases diagnosed from 1985-94, the
5-year survival rate for Hodgkin’s disease
was 91% for children younger than 20
years of age (Figure II.8). White children
had a slight survival advantage over black
children (92% versus 84% 5-year survival),
and males and females had similar out-
come (90% versus 92% 5-year survival).
Outcomes were also similar for children
diagnosed younger than 15 years of age
compared to 15-19 year olds (92% versus
90% 5-year survival).
RISK FACTORS (TABLE II.4)
Available epidemiological and molecu-
lar biological data suggest that Hodgkin’s
disease among children and adolescents
represents at least two distinctive condi-
tions [14-16]. In one condition, EBV ge-
nomic sequences are typically present in
the Reed-Sternberg cells. This form of
Hodgkin’s disease is more common for

41
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
Figure II.8: Hodgkin's disease 5 year-relative survival rates
by sex, race, age and time period
SEER (9 areas), 1975-84 and 1985-94
87 87 87 87
86
83
88
91
90
92 92
84
92
91
Total Male Female White Black <5 5-10 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race Age
#
#
# - <25 cases - rate not shown
Table II.4: Current knowledge on causes of Hodgkin’s disease in children
Exposure or Characteristic Comments References
Family history of Hodgkin’s
disease
Monozygotic twins of young adult patients have a 99-fold increased risk.
Other siblings have a 7-fold increased risk.
22,33,34
Epstein-Barr virus infection EBV-associated HD is associated with mixed cellularity vs. nodular
sclerosis histologic subtypes, children from economically less-developed
vs. more-developed regions and young adult males vs. females.
Additionally, history of infectious mononucleosis and high titer
antibodies to EBV are associated with HD among young adults, although
paradoxically HD cases among the young adult population typically do
not have detectable EBV genomic sequences in tumor tissue.
7,8,16-18,20,21,35-37
Socioeconomic status For young adult disease (ages 16-44), risk increases with socioeconomic
status and with related characteristics such as small family size and
single family housing. In children younger than 10 years of age, risk
appears higher for lower socioeconomic status.
15,38-40
Social contacts For young adult disease, having fewer siblings and childhood playmates
is associated with higher risk. These findings suggest that infections in
early childhood may reduce risk of young adult disease.
19,33
Clustering Young adult cases (age 16-45 years) tend to cluster more than older cases
(age 46-70).
41
Known to increase risk
Factors for which evidence is
inconsistent or limited

42
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
childhood cases occurring in developing
countries compared to cases occurring in
countries with higher socioeconomic status
[7,8]. EBV involvement is also associated
with male sex, mixed cellularity subtype,
and young age at diagnosis [7,8,16-18].
However, the associated environmental
and/or genetic factors that cause EBV
infection, which is common among young
children (particularly in developing coun-
tries), to result in Hodgkin’s disease in a
very small percentage of children are not
understood. A second form of Hodgkin’s
disease is primarily of the nodular sclerosis
subtype, arising in older adolescents and
young adults living in more affluent societ-
ies [15,16]. In spite of observations sug-
gesting a relationship between Hodgkin’s
disease among older adolescents/young
adults and infectious mononucleosis and
high titers of antibodies against EBV [19-
21], cases of Hodgkin’s disease in this
population typically do not have EBV
genomic sequences detectable in tumor
tissue [7,8].
For Hodgkin’s disease arising in young
adults, genetic susceptibility may be a
factor for some cases, based on the greatly
increased relative risk for Hodgkin’s dis-
ease occurring in both members of monozy-
gotic twin pairs compared to the risk of
occurrence in dizygotic twin pairs [22].
Further support for genetic predisposition
to Hodgkin’s disease is the consistently low
incidence of Hodgkin’s disease (especially of
the nodular sclerosis subtype) among
populations of East Asian ethnic origin and
the high incidence (especially of mixed
cellularity subtype) among some popula-
tions of South Asian origin, with these
differences appearing to be independent of
socioeconomic status [15].
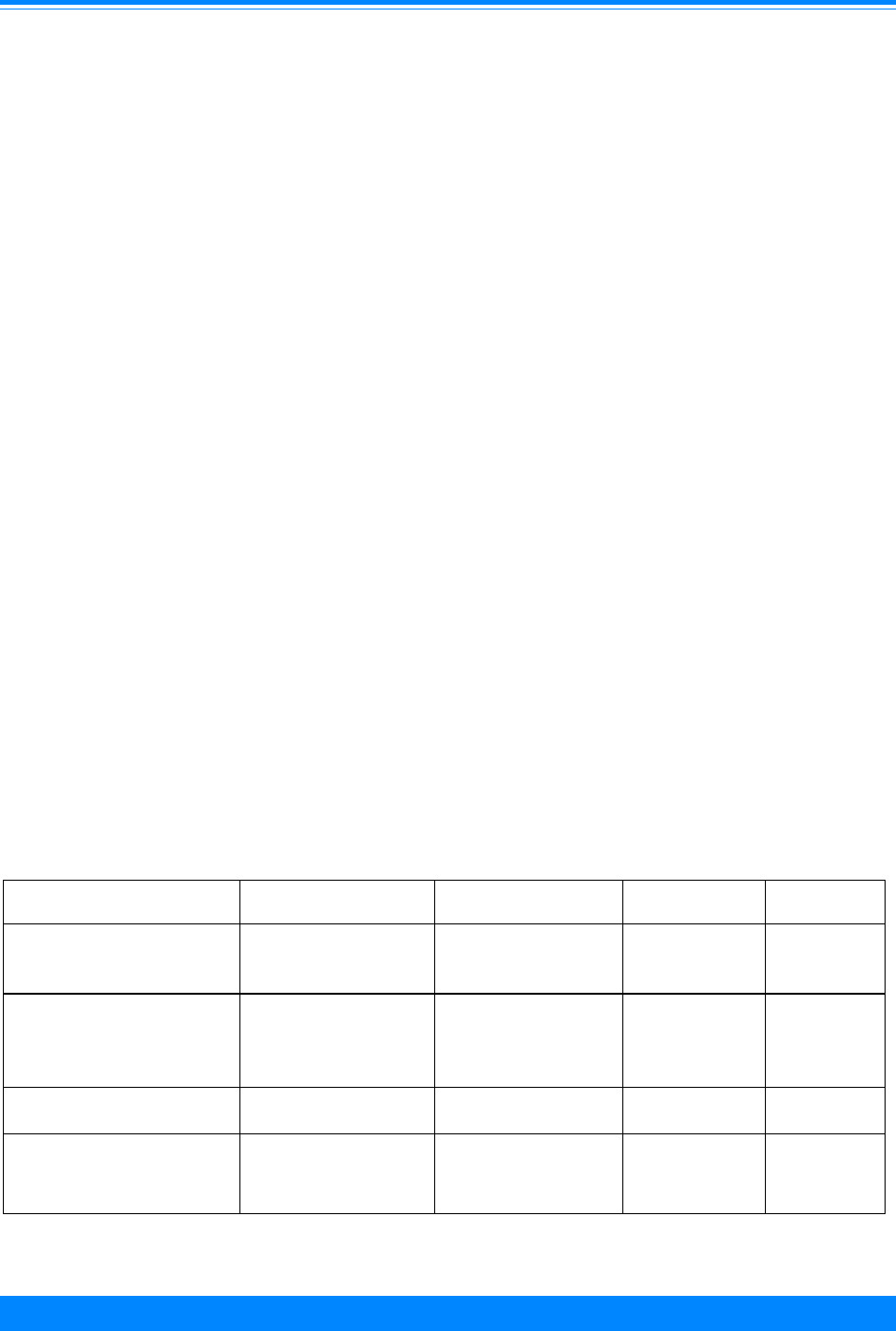
43
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
(but in the jaws of young children in Af-
rica), and with lymphoblastic lymphomas
commonly present as large mediastinal
masses. The lymphoblastic lymphomas of
children are indistinguishable histologically
from T-cell acute lymphoblastic leukemia
(ALL). The clinical and biological charac-
teristics of each of these categories of
childhood NHL are summarized in Table
II.5.
Histologic subtype
Figure II.9 illustrates the distinctive
distributions by age and by sex of the NHL
subtypes that predominate in US children
in the geographic areas covered by the
SEER Program. For Burkitt’s and
Burkitt’s-like lymphoma, the incidence was
much higher for males than for females.
Five to fourteen year olds had higher rates
than children younger than 5 years of age
and adolescents 15-19 years of age. For
children younger than 20 years of age, the
incidence for the Burkitt’s lymphomas was
nearly 5.0-fold higher for males (3.2 per
million) than for females (0.7 per million).
NON-HODGKIN’S LYMPHOMA
Table II.5: Major histopathological categories of non-Hodgkin’s lymphoma in children*
Category (REAL) Category (Working
Formulation)
Clinical Presentation Chromosomal
Translocation
Genes
Affected
Burkitt’s and Burkitt-like
lymphomas
ML small noncleaved cell Intraabdominal
(sporadic), jaw (endemic)
t(8;14)(q24;q32)
t(2;8)(p11;q24)
t(8;22)(q24;q11)
C-MYC, IgH
IgK,Ig
l
Lymphoblastic lymphoma,
precursor T
Lymphoblastic
convoluted and
nonconvoluted
Mediastinal MTS1/p16ink4a
Deletion TAL1
t(1;14)(p34;q11)
t(11;14)(p13;q11)
TAL1, TCRad,
RHOMB1,
HOX11
Anaplastic large cell lymphoma ML immunoblastic or ML
large cell
Variable t(2;5)(p23;q35) ALK, NMP
Diffuse large cell lymphoma ML large cell Variable t(8;14)(q24;q32) in
adults, but not
well characterized
in children
BCL2, IgH (in
adults)
* Adapted from Shad and Magrath [31], and from Goldsby and Carroll [6].
The non-Hodgkin’s lymphomas that
develop in children are distinct from the
more common forms of lymphomas in
adults. While the lymphomas in adults are
more commonly of low or intermediate
grade, almost all of those that occur in
children can be classified into one of four
high-grade categories: 1) the Burkitt’s and
Burkitt-like lymphomas (small noncleaved
cell lymphomas); 2) lymphoblastic lympho-
mas; 3) diffuse large B-cell lymphomas;
and 4) anaplastic large cell lymphomas.
Each of these types of childhood NHL is
associated with distinctive molecular
biological characteristics, with transloca-
tions resulting in activation of the C-MYC
gene occurring in the Burkitt’s and Burkitt-
like lymphomas, translocations involving
the TAL1 gene and the T-cell receptor genes
typifying the lymphoblastic lymphomas,
and translocations involving the NPM gene
at chromosome band 5q35 in the anaplastic
large cell lymphomas. These NHL catego-
ries are also associated with characteristic
clinical presentations, with the Burkitt’s
and Burkitt-like lymphomas commonly
present in the abdomen among US children

44
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
In contrast to the Burkitt’s lymphomas, the
incidence of diffuse large cell lymphoma
rose steadily with increasing age, with
males showing somewhat higher incidence
than females for the population younger
than 20 year age group (M/F = 1.4). Lym-
phoblastic lymphoma occurred at similar
frequency across all 5-year age groups, and
like the other lymphoma subtypes for
children younger than 20 years of age was
more common in males than females (M/F
= 2.5).
Age-specific incidence
The contribution of the non-Hodgkin’s
lymphomas to the overall cancer incidence
increased from 3% for children younger
than 5 years of age to 8-9% for 10-14 and
15-19 year olds (Figure II.1). Figure II.3
illustrates that the incidence of NHL varied
much less by age than did Hodgkin’s dis-
ease. The incidence of NHL rapidly in-
creased in the first three years of life,
before reaching a plateau rate of approxi-
mately 10 per million at around 4 years of
age (Figure II.4). A similar age-incidence
Figure II.10: NHL age-specific incidence
rates by sex, all races, SEER
1976-84 and 1986-94 combined
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
01234567891011121314151617181920
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
Male
Female
"
)
Figure II.9: NHL age-specific incidence rates by histologic group
sex, and age, all races, SEER (9 areas), 1977-95
0.6
1
1.9
5.5
0.5
1.2
2.5
6.1
0.6
0.7
1.4
4.9
1.3
1.6
1.8
1.6
1.6
2.2
2.8
2.2
0.9
1
0.7
0.9
2
3.6
3.5
2
3.2
6
6.1
2.8
0.8
1.1
0.8
1.2
1.9
2.5
3.6
5.6
2.3
3.3
4.3
7.8
1.5
1.6
2.8
3.4
<5 5-9 10-14 15-19 <5 5-9 10-14 15-19 <5 5-9 10-14 15-19
0
5
10
15
20
Average annual rate per million
Diffuse large cell
Lymphoblastic
Burkitt's
Other
Both sexes
Male
Female

45
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
pattern was observed for males and fe-
males (Figure II.10), but the incidence rate
among children older than 4 years of age
was much higher for males than for fe-
males. While incidence rates for NHL
remained fairly stable through the remain-
der of the first decade, the incidence began
increasing after age 10 (Figures II.4 and
II.10). Part of this increase in incidence
among adolescents was due to higher rates
for diffuse large cell lymphomas compared
to rates for this subtype in younger chil-
dren (Figure II.9).
Sex-specific incidence
There was a notable male predomi-
nance for NHL in children, with 70% of the
cases occurring in males (Table II.1). The
male predominance was seen for all age
groups (Figures II.10 and II.11), although
it was more pronounced for children
younger than 15 years of age (M/F = 3.0)
Figure II.11: NHL age-specific and age-adjusted*
incidence rates by age and sex with male to female
ratios (M/F), all races, SEER, 1990-95
9
12.2
16.1
20.5
12.7
14.7
3.6
3.9
5
11.9
4.2
6.1
<5 5-9 10-14 15-19 <15 <20
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
Male
Female
M/F=2.5
M/F=3.1
M/F=3.2
M/F=1.7
M/F=3.0
M/F=2.4
*Adjusted to the 1970 US standard population
Figure II.12: NHL age-specific and age-adjusted* incidence rates by age
and race with white to black ratios (W/B) both sexes, SEER, 1990-95
5.9
9.4
10.6
16.1
8.9
10.6
5.1
4.8
10.2
9.4
6.8
7.5
<5 5-9 10-14 15-19 <15 <20
Age (in years) at diagnosis
0
5
10
15
20
Average annual rate per million
White
Black
*Adjusted to the 1970 US standard population
W/B=2.0
W/B=1.0
W/B=1.7
W/B=1.4
W/B=1.3
W/B=1.2

46
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
compared to 15-19 year olds (M/F = 1.7).
As noted in the preceding paragraph, the
age-incidence pattern is similar for males
and females, although incidence is higher
at all age groups for males than females
(Figure II.10).
Black-white difference in incidence
The incidence of NHL among white
children was 1.4-fold and 1.3-fold higher
than that for black children for the younger
than 15 years of age group and younger
than 20 years of age group, respectively
(Figure II.12). The difference in incidence
between white and black children appeared
greatest for children 5-9 years of age and
15-19 years of age (Figure II.12).
Trends in incidence rates
The incidence of NHL remained stable
for children younger than 15 years of age
from 1975 through 1995 (Table II.3). How-
Figure II.13: Trends in NHL age-adjusted*
incidence rates, all races
both sexes, SEER, 1975-95
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
1975 1980 1985 1990 1995
Year of diagnosis
0
5
10
15
20
25
Average annual rate per million
<15 Years
15-19 Years
(
&
*Adjusted to the 1970 US standard population
Figure II.14: NHL 5-year relative survival rates by sex, race, age
and time period, SEER (9 areas), 1975-84 and 1985-94
56
55
59
55
53
48
60
57
56
72 72 72
73
72
75
77
71
69
Total Male Female White Black <5 5-10 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex Race
Age

47
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
ever, the incidence among 15-19 year olds
increased from 10.7 per million (1975-79) to
16.3 per million (1990-95) (Figure II.13).
This increase among the older adolescents/
young adults is similar to that reported for
young adults older than 20 years of age
[13,23].
SURVIVAL
The 5-year survival rate for children
younger than 20 years of age with NHL
was 72% for the years 1985-94, a substan-
tial improvement from the 56% survival
rate for the years 1975-84 (Figure II.14).
For the most recent 10-year period, survival
was similar for white and black children
and was similar for males and females.
During this 10-year period, children
younger than 10 years of age had slightly
better 5-year survival rates than did those
10-19 years of age (approximately 76%
versus 70%).
RISK FACTORS (TABLE II.6)
The etiology of most cases of childhood
NHL is unknown. In a small proportion of
cases, childhood NHL is linked with various
disorders of immune dysfunction. Congeni-
tal immunodeficiency syndromes (e.g.,
Wiskott-Aldrich, ataxia-telangiectasia, X-
linked lymphoproliferative syndrome, and
severe combined immunodeficiency) and
acquired immunodeficiency syndrome
(AIDS) from human immunodeficiency
virus (HIV) infection are associated with an
increased risk of NHL [24-26]. Persons who
are immuno-compromised as a result of
organ and bone marrow transplantation
are also at increased risk of NHL
[24,27,28]. EBV is associated with the
endemic or ‘African-type’ Burkitt’s lympho-
mas, but much less commonly with the
sporadic Burkitt’s lymphomas in the US
[29-31]. Few epidemiologic studies of
childhood cancer have focused exclusively
on NHL, and most prenatal and perinatal
exposures evaluated to date were not
associated with increased or decreased risk
[32].
SUMMARY
Overall, age-adjusted incidence rates
for Hodgkin’s disease and NHL were simi-
lar for children and adolescents younger
than 20 years of age, although the age-
specific incidence patterns are markedly
different. Among young children, Hodgkin’s
disease is more common among males than
females, whereas for older adolescents
Hodgkin’s disease is more common among
females. Hodgkin’s disease has a high 5-
year relative survival rate, currently
greater than 90%. For unknown reasons,
Table II.6: Current knowledge on causes of non-Hodgkin’s lymphoma (NHL) in children
Exposure or Characteristic Comments References
Immunodeficiency Immunosuppressive therapy, congenital immunodeficiency syndromes (e.g.,
ataxia telangiectasia), acquired immunodeficiency syndrome (AIDS) all
predispose to NHL.
24-27,42,43
Epstein-Barr virus EBV is associated with ‘African-type’ Burkitt’s lymphoma, and chronic
immune suppression due to malaria may be a co-factor in this situation. EBV
is also associated with NHL in patients with immunodeficiency.
27,29-31,44,45
Radiation While a few studies report increased NHL risk in adults or children with
ionizing or electromagnetic field (EMF) radiation, others report no association.
46-50
Known risk factors
Substantial evidence
implicating factor
Factors for which evidence is
inconsistent or limited

48
National Cancer Institute
SEER Pediatric Monograph
ICCC II LYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
the incidence of Hodgkin’s disease appears
to be slowly decreasing for both younger
children and for older adolescents. Sub-
stantial evidence implicates EBV in the
etiology of a subset of Hodgkin’s disease
(primarily mixed cellularity subtype), but
the mechanism by which EBV results in
development of Hodgkin’s disease and the
potential role of co-factors are not under-
stood.
The non-Hodgkin’s lymphomas of
children are a heterogeneous group of
tumors, with Burkitt’s and Burkitt-like
tumors predominating among 5-14 year
olds, and with diffuse large cell lymphomas
being the most common subtype among 15-
19 year olds. Particularly for the Burkitt’s
lymphomas, there is a marked sex differen-
tial, with males having a much higher
incidence than females. Survival rates
have improved substantially for NHL
between the late 1970s and the late 1980s
are now over 70%, with similar rates for
both sexes and for whites and blacks. The
incidence of NHL among children younger
than 15 years of age appears fairly con-
stant over the past 21 years, while there
appears to have been a slight increase in
incidence for the 15-19 year old age group.
The etiology of childhood NHL is poorly
understood, although a small proportion of
cases arise in children with congenital or
acquired severe immune dysfunction.
Reference List
1. Parkin D, Stiller C, Draper G, et al: Interna-
tional Incidence of Childhood Cancer. Lyon:
World Health Organization, 1988.
2. Kuppers R, Hansmann ML, Rajewsky K:
Clonality and germinal centre B-cell derivation
of Hodgkin/Reed- Sternberg cells in Hodgkin’s
disease. Ann Oncol 9:S17-20, 1998.
3. Izban KF, Nawrocki JF, Alkan S, et al: Mono-
clonal IgH gene rearrangement in
microdissected nodules from nodular sclerosis
Hodgkin disease. Am J Clin Pathol 110:599-
606, 1998.
4. Vockerodt M, Soares M, Kanzler H, et al:
Detection of clonal Hodgkin and Reed-
Sternberg cells with identical somatically
mutated and rearranged VH genes in different
biopsies in relapsed Hodgkin’s disease. Blood
92:2899-907, 1998.
5. Irsch J, Nitsch S, Hansmann ML, et al: Isola-
tion of viable Hodgkin and Reed-Sternberg
cells from Hodgkin disease tissues. Proc Natl
Acad Sci U S A 95:10117-22, 1998.
6. Goldsby RE, Carroll WL: The molecular biology
of pediatric lymphomas. J Pediatr Hematol
Oncol 20:282-96, 1998.
7. Sleckman BG, Mauch PM, Ambinder RF, et al:
Epstein-Barr virus in Hodgkin’s disease:
correlation of risk factors and disease charac-
teristics with molecular evidence of viral
infection. Cancer Epidemiol Biomarkers Prev
7:1117-21, 1998.
8. Glaser SL, Lin RJ, Stewart SL, et al: Epstein-
Barr virus-associated Hodgkin’s disease:
epidemiologic characteristics in international
data. Int J Cancer 70:375-82, 1997.
9. Hudson M, Donaldson S: Hodgkin’s disease. In
Principles and Practice of Pediatric Oncology
(Pizzo P, Poplack D, eds). Philadelphia:
Lippincott-Raven Publishers, 1997, pp 523-543.
10. Harris NL: The many faces of Hodgkin’s
disease around the world: what have we
learned from its pathology? Ann Oncol 9:S45-
56, 1998.
11. Franklin J, Tesch H, Hansmann ML, et al:
Lymphocyte predominant Hodgkin’s disease:
pathology and clinical implication. Ann Oncol
9:S39-44, 1998.
12. Cartwright R, McNally R, Roman E, et al:
Incidence and time trends in Hodgkin’s
disease: from parts of the United Kingdom
(1984-1993) [In Process Citation]. Leuk
Lymphoma 31:367-77, 1998.
13. Hartge P, Devesa SS, Fraumeni JF, Jr.:
Hodgkin’s and non-Hodgkin’s lymphomas.
Cancer Surv 20:423-53, 1994.
14. MacMahon B: Epidemiology of Hodgkin’s
disease. Cancer Res 26:1189-201, 1966.
15. Stiller CA: What causes Hodgkin’s disease in
children? Eur J Cancer 34:523-8, 1998.
16. Armstrong AA, Alexander FE, Cartwright R, et
al: Epstein-Barr virus and Hodgkin’s disease:
further evidence for the three disease hypoth-
esis. Leukemia 12:1272-6, 1998.
17. Armstrong AA, Alexander FE, Paes RP, et al:
Association of Epstein-Barr virus with pediat-
ric Hodgkin’s disease. Am J Pathol 142:1683-8,
1993.
18. Kusuda M, Toriyama K, Kamidigo NO, et al: A
comparison of epidemiologic, histologic, and
virologic studies on Hodgkin’s disease in
western Kenya and Nagasaki, Japan. Am J
Trop Med Hyg 59:801-7, 1998.
19. Gutensohn N, Cole P: Childhood social environ-
ment and Hodgkin’s disease. N Engl J Med
304:135-40, 1981.

49
National Cancer Institute SEER Pediatric Monograph
ICCC IILYMPHOMAS AND RETICULOENDOTHELIAL NEOPLASMS
20. Evans AS, Gutensohn NM: A population-based
case-control study of EBV and other viral
antibodies among persons with Hodgkin’s
disease and their siblings. Int J Cancer 34:149-
57, 1984.
21. Mueller N, Evans A, Harris NL, et al:
Hodgkin’s disease and Epstein-Barr virus.
Altered antibody pattern before diagnosis. N
Engl J Med 320:689-95, 1989.
22. Mack TM, Cozen W, Shibata DK, et al: Concor-
dance for Hodgkin’s disease in identical twins
suggesting genetic susceptibility to the young-
adult form of the disease [see comments]. N
Engl J Med 332:413-8, 1995.
23. Devesa SS, Blot WJ, Stone BJ, et al: Recent
cancer trends in the United States. J Natl
Cancer Inst 87:175-82, 1995.
24. Kersey JH, Shapiro RS, Filipovich AH: Rela-
tionship of immunodeficiency to lymphoid
malignancy. Pediatr Infect Dis J 7:S10-2, 1988.
25. Filipovich AH, Mathur A, Kamat D, et al:
Primary immunodeficiencies: genetic risk
factors for lymphoma. Cancer Res 52:5465s-
5467s, 1992.
26. McClain KL, Joshi VV, Murphy SB: Cancers in
children with HIV infection. Hematol Oncol
Clin North Am 10:1189-201, 1996.
27. Hanto DW, Najarian JS: Advances in the
diagnosis and treatment of EBV-associated
lymphoproliferative diseases in
immunocompromised hosts. J Surg Oncol
30:215-20, 1985.
28. Penn I: The changing pattern of posttransplant
malignancies. Transplant Proc 23:1101-3, 1991.
29. de-The G, Geser A, Day NE, et al: Epidemio-
logical evidence for causal relationship be-
tween Epstein-Barr virus and Burkitt’s
lymphoma from Ugandan prospective study.
Nature 274:756-61, 1978.
30. Gutierrez MI, Bhatia K, Barriga F, et al:
Molecular epidemiology of Burkitt’s lymphoma
from South America: differences in breakpoint
location and Epstein-Barr virus association
from tumors in other world regions. Blood
79:3261-6, 1992.
31. Shad A, Magrath I: Malignant non-Hodgkin’s
lymphomas in children. In Principles and
Practice of Pediatric Oncology (Pizzo P, Poplack
D, eds). Philadelphia: Lippincott-Raven
Publishers, 1997, pp 545-587.
32. Adami J, Glimelius B, Cnattingius S, et al:
Maternal and perinatal factors associated with
non-Hodgkin’s lymphoma among children. Int
J Cancer 65:774-7, 1996.
33. Grufferman S, Cole P, Smith PG, et al:
Hodgkin’s disease in siblings. N Engl J Med
296:248-50, 1977.
34. Ferraris AM, Racchi O, Rapezzi D, et al:
Familial Hodgkin’s disease: a disease of young
adulthood? Ann Hematol 74:131-4, 1997.
35. Ambinder R, Browning P, Lorenzana I, et al:
Epstein-Barr virus and childhood Hodgkin’s
disease in Honduras and the United States.
Blood 81:462-7, 1993.
36. Chang KL, Albujar PF, Chen YY, et al: High
prevalence of Epstein-Barr virus in the Reed-
Sternberg cells of Hodgkin’s disease occurring
in Peru. Blood 81:496-501, 1993.
37. Pallesen G, Hamilton-Dutoit SJ, Rowe M, et al:
Expression of Epstein-Barr virus latent gene
products in tumour cells of Hodgkin’s disease
[see comments]. Lancet 337:320-2, 1991.
38. Grufferman S, Delzell E: Epidemiology of
Hodgkin’s disease. Epidemiol Rev 6:76-106,
1984.
39. Gutensohn NM: Social class and age at
diagnosis of Hodgkin’s disease: new epidemio-
logic evidence for the “two-disease hypothesis”.
Cancer Treat Rep 66:689-95, 1982.
40. Gutensohn NM, Shapiro DS: Social class risk
factors among children with Hodgkin’s disease.
Int J Cancer 30:433-5, 1982.
41. Greenberg RS, Grufferman S, Cole P: An
evaluation of space-time clustering in Hodgkin’s
disease. J Chronic Dis 36:257-62, 1983.
42. Spector BD, Perry GSd, Kersey JH: Genetically
determined immunodeficiency diseases (GDID)
and malignancy: report from the immunodefi-
ciency—cancer registry. Clin Immunol
Immunopathol 11:12-29, 1978.
43. Taylor AM, Metcalfe JA, Thick J, et al: Leuke-
mia and lymphoma in ataxia telangiectasia.
Blood 87:423-38, 1996.
44. Niedobitek G, Young LS, Herbst H: Epstein-
Barr virus infection and the pathogenesis of
malignant lymphomas. Cancer Surv 30:143-62,
1997.
45. McClain K, Wu W-S, Joshi VV, et al: Lympho-
mas and B-cell leukemias of children with
AIDS are infrequently associated with muta-
tions or translocations of c-myc or with Epstein
Barr virus. Blood 88:A1505, 1996.
46. Boice JD, Jr.: Radiation and non-Hodgkin’s
lymphoma. Cancer Res 52:5489s-5491s, 1992.
47. Pearce N, Bethwaite P: Increasing incidence of
non-Hodgkin’s lymphoma: occupational and
environmental factors. Cancer Res 52:5496s-
5500s, 1992.
48. Tynes T, Haldorsen T: Electromagnetic fields
and cancer in children residing near Norwe-
gian high-voltage power lines. Am J Epidemiol
145:219-26, 1997.
49. Verkasalo PK, Pukkala E, Hongisto MY, et al:
Risk of cancer in Finnish children living close
to power lines [see comments]. BMJ 307:895-9,
1993.
50. Olsen JH, Nielsen A, Schulgen G: Residence
near high voltage facilities and risk of cancer
in children [see comments]. BMJ 307:891-5.

50
National Cancer Institute
SEER Pediatric Monograph

ICCC IIICNS AND MISCELLANEOUS INTRACRANIAL AND INTRASPINAL NEOPLASMS
51
National Cancer Institute
SEER Pediatric Monograph
HIGHLIGHTS
Incidence
♦ The CNS malignancies represented 16.6% of all malignancies during childhood
(including adolescence). CNS cancer as a group was the second most frequent
malignancy of childhood and the most common of the solid tumors. In the US
approximately 2,200 children younger than 20 years of age are diagnosed annually
with invasive CNS tumors.
♦ Astrocytomas accounted for 52% of CNS malignancies, PNET comprised 21%,
other gliomas 15% and ependymomas an additional 9% (Figure III.1).
♦ Unlike adults and older children, young children have a relatively high occurrence
of malignancies in the cerebellum and the brain stem. In fact, in children younger
than 10 years of age, brain stem malignancies were nearly as common as cerebral
malignancies, and cerebellum malignancies were far more common than cerebral
malignancies (Figure III.2).
♦ The incidence of invasive CNS tumors was higher in males than females and
higher among white children than black children (Figure III.5).
♦ The average annual incidence of CNS cancer varied only slightly by age of diagno-
sis from infancy (36.2 per million) through age 7 years (35.2 per million). From
age 7 to 10, a 40% drop in the incidence rate (to 21.0 per million) was observed.
CNS cancer rates were fairly consistent among children aged 11 through 17 years,
until another substantial decrease occurred at age 18 (Figure III.6).
♦ The increase in CNS cancer rates in the past two decades has been the subject of
numerous reports. One concern is that changes in environmental exposures may
be responsible for the increasing incidence rates, although epidemiologic evidence
to support this hypothesis currently is lacking. An alternative explanation is that
improvements in diagnostic technology and case ascertainment may be
contributing to the increasing trend.
Survival
♦ In general, children with CNS cancer do not share the favorable prognosis of those
with many other common pediatric neoplasms.
♦ Very young children with CNS cancer, especially infants with ependymoma or
PNET, had low survival rates (Table III.2).
Risk factors
♦ There is no specific risk factor that explains a substantial proportion of brain
tumor occurrence, but there are a couple of factors that explain a small proportion
(Table III.3).
INTRODUCTION
Since most of the neoplasms described
in this chapter are in the central nervous
system, the abbreviation CNS will be used
to refer to neoplasms that originate in the
brain, other intracranial sites such as the
pituitary or pineal glands, and the spinal
cord. In the US, approximately 2,200
children and adolescents younger than 20
years of age are diagnosed with malignant
central nervous system tumors each year.
Over 90 percent of primary CNS malignan-
cies in children are located within the
James G. Gurney, Malcolm A. Smith, Greta R. Bunin

ICCC III CNS
52
National Cancer Institute
SEER Pediatric Monograph
rates for the CNS germ cell malignancies
from 1990-95 were 0.2 per million children
younger than 15 years of age, and 1.9 per
million children younger than 20 years of
age. Fifty-three additional tumors were
excluded because they occurred outside the
brain, intracranium and spinal cord.
It also should be noted that data
reported here are comprised solely of CNS
tumors that are classified as primary and
malignant. Primary CNS neoplasms are
tumors that originated in the central
nervous system. Thus, they exclude cancer
that developed in some other location in the
body and then spread to the CNS. Like-
wise, CNS tumors classified as benign or
with uncertain behavior (nonmalignancies)
are not routinely collected by SEER areas,
and thus are not included in this report.
The pathological distinction between malig-
nant and nonmalignant tumors of the CNS
is not always consistent with clinical behav-
ior, particularly for intracranial tumors.
Depending on the location and the size of
the tumor, some intracranial tumors that
are classified as benign can have a destruc-
tive clinical course (eg. craniopharyngioma).
In contrast, some tumors classified as
malignant may require no treatment and
have little clinical significance (eg. pilocytic
astrocytomas of the optic pathway). Al-
though all central registries will include
malignant neoplasms in their case ascer-
tainment, when comparing CNS incidence
rates across cancer surveillance systems it
is necessary to determine whether a given
registry also includes nonmalignant tu-
mors. An analysis of data from the Central
Brain Tumor Registry of the United States
(a compilation of data from population-
based registries that include case ascertain-
ment of nonmalignant CNS tumors)
showed that the incidence of only malig-
nant CNS tumors underestimates the
incidence of both malignant and non-
malignant CNS tumors by approximately
28% [4].
brain. This report only includes malignant
CNS tumors.
Classification system
CNS tumors are heterogeneous in
regards to histology and clinical course.
Because of the many relatively similar
histopathological types and their rarity, it is
necessary for epidemiologic purposes to
group CNS tumors into rather broad histo-
logic categories. There are several classifi-
cation systems that are used for describing
CNS tumors and no system has yet
emerged as the definitive gold standard
[1,2]. For most of this monograph, malig-
nancies are grouped according to the Inter-
national Classification of Childhood Cancer
(ICCC) system [3]. There are a few minor
discrepancies within the ICCC system for
CNS tumors, however, that somewhat
compromise accurate comparisons with
other published work. Most notable, intrac-
ranial neuroblastoma and pineoblastoma,
which, along with medulloblastoma are
generally considered primitive neuroecto-
dermal tumors (PNET), are not included
with the PNET category of the ICCC for
CNS. For the descriptive analysis that
follows, we modified the ICCC groupings for
CNS tumors in the following manner:
“Other specified intracranial and intraspi-
nal neoplasms excluding pineoblastoma
(IIIe)” and “Unspecified intracranial and
intraspinal neoplasms (IIIf)” were com-
bined into one category, called ‘other CNS’;
the “Ependymoma (IIIa)” category was not
changed; the “PNET (IIIc)” category was
expanded to include intracranial neuroblas-
toma (these were also reported with ICCC
IV) and pineoblastoma. Finally, the ICCC
system places intracranial and intraspinal
germ cell malignancies within the germ cell
category, rather than the CNS tumor
category. We chose to follow the ICCC
system for CNS germ cell tumors, thus we
did not include intracranial and intraspinal
germ cell tumors in this chapter (see ICCC
group X). The average annual incidence

ICCC IIICNS
53
National Cancer Institute SEER Pediatric Monograph
INCIDENCE
Unless otherwise indicated, the discus-
sion on incidence that follows will pertain
to children younger than 20 years of age
and only malignant tumors. For the 21-
year period of 1975-95, there were 4,945
primary malignant tumors of the CNS
diagnosed among children in SEER areas.
This represented 16.6% of all malignancies
during childhood (including adolescence).
CNS cancer as a group was the second most
frequent malignancy of childhood and the
most common of the solid tumors. Astrocy-
tomas accounted for 52% of CNS malignan-
cies, PNET comprised 21%, other gliomas
15%, and ependymomas an additional 9%
(Figure III.1).
The incidence rates by location within
the brain and other CNS sites as a function
of age are shown in Figure III.2. Unlike
adults and older children, who have higher
rates in the cerebrum, young children have
a relatively high occurrence of malignancies
in the cerebellum and the brain stem. In
fact, in children between the ages of 5 and
9, brain stem malignancies were nearly as
common as cerebral malignancies, and
cerebellum malignancies were far more
common than cerebral malignancies. The
pattern shifted among children between the
ages of 10-19, in that the incidence of both
brain stem and cerebellar cancers de-
creased while cerebral malignancies in-
creased slightly. The “other” brain site
group included the ventricles, where
ependymomas generally develop, and
malignancies with brain sites not otherwise
specified. The “Other CNS” category in-
cludes malignancies of the meninges,
cranial nerves and spinal cord.
Figure III.1: Percent distribution of malignant CNS
tumors by age and histologic group, all races
both sexes, SEER, 1975-95
49.6
22.9
15.4
9.3
2.7
52.2
20.8
15.5
8.6
3
Astrocytomas
PNET
Other gliomas
Ependymomas
Other CNS
010203040506070 0 10203040506070
<15 years
<20 years
Percent of total CNS cancer
Figure III.2: Malignant CNS tumor age-specific
incidence rates by anatomic site and age
all races, both sexes, SEER, 1975-95
4.7
5.9
2.8
1.7
5.8
5.9
6.8
7
9.3
9.7
5.7
3.7
8.8
5.9
4.5
4
2.6
1.9
1.7
1.6
<5
5-9
10-14
15-19
Age (in years) at diagnosis
0123456789101112
Average annual rate per million
Brain Stem
Cerebrum
Cerebellum
Other Brain
Other CNS
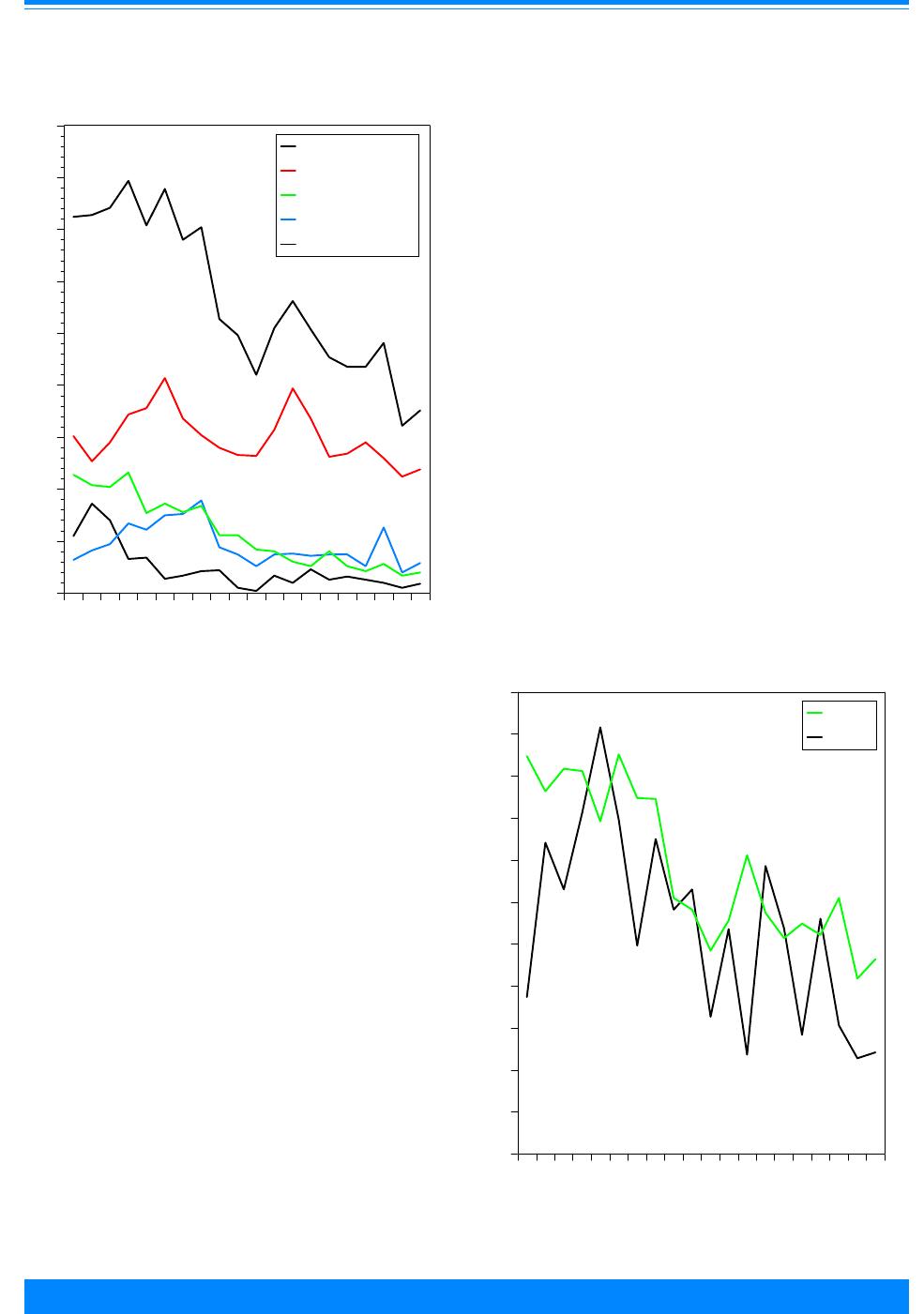
ICCC III CNS
54
National Cancer Institute
SEER Pediatric Monograph
Age-specific incidence
Incidence rates by single year of age
are presented in Figure III.3.
1
The average
annual incidence of CNS cancer varied only
slightly by age of diagnosis from infancy
(36.2 per million) through age 7 years (35.2
per million). From age 7 to 10, a 40% drop
in the incidence rate (to 21.0 per million)
was observed. CNS cancer rates were
fairly consistent among children aged 11
through 17 years, until another substantial
decrease occurred at age 18.
The incidence of astrocytomas peaked
at age 5 (20.7 per million) and a second
peak occurred at age 13 (19.7 per million).
PNET rates were fairly steady from infancy
through age 3 years (ranging from 11.6 to
10.2 per million) and then steadily declined
thereafter. Rates of ependymomas were
highest through age 3 years, with the age
of peak incidence occurring during the
second year of life (8.6 per million). Among
children aged 5-14, ependymomas are very
rare, averaging only 1.4 per million.
Although in our data the age-specific
rates for black children were fairly unstable
because of small numbers of cases (295
cases from 1986-94), the greatest difference
in rates between whites and blacks was
observed during the first year of life (47.8
vs. 18.7 per million, respectively) (Figure
III.4). In the second year of life, rates
among whites decreased from the first year,
Figure III.3: Malignant CNS tumor age-specific
incidence rates, all races, both sexes
SEER, 1986-94
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
&
&
&
(
(
(
(
(
(
(
(
(
((
(
(
(
(
(
(
(
(
(
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
##
#
#
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
40
45
Average annual rate per million
All CNS
Astrocytomas
PNET
Other gliomas
Ependymoma
#
$
(
&
)
Figure III.4: Malignant CNS tumor age-specific
incidence rates by race, both sexes
SEER, 1986-94
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
01234567891011121314151617181920
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
40
45
50
55
Average annual rate per million
White
Black
+
'
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.
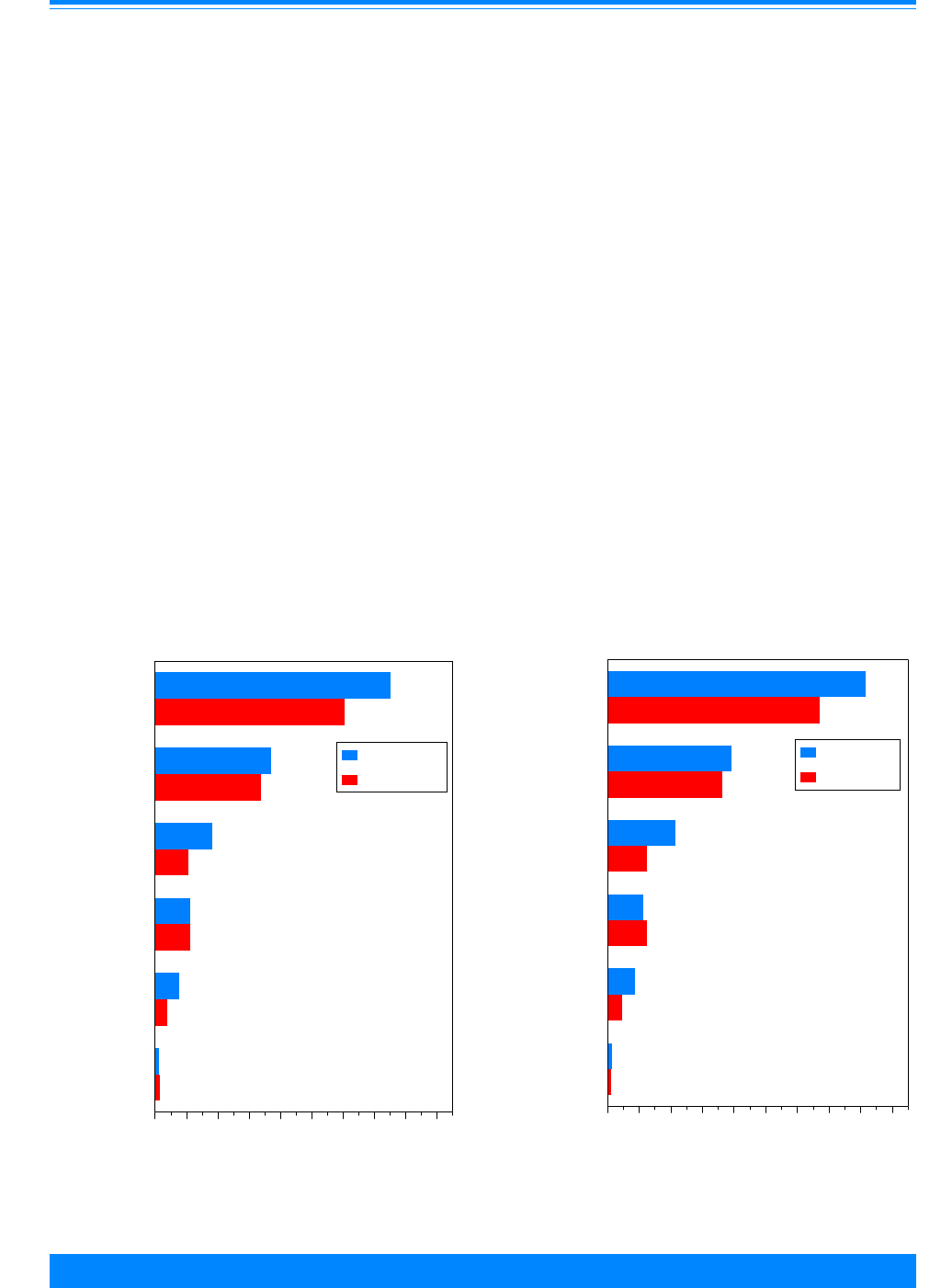
ICCC IIICNS
55
National Cancer Institute SEER Pediatric Monograph
while rates in blacks increased substan-
tially. To a degree, this could suggest a
pattern in which whites were diagnosed
earlier than blacks (on average) for the
CNS malignancies that occur early in life,
although we are aware of no other evidence
that supports this speculation.
Sex-specific incidence
As will be discussed below, brain cancer
incidence rates in children have increased
in SEER areas over the past 2 decades. For
this reason, the following CNS cancer
incidence rates are reported for the time
period 1990-95, rather than 1975-95, to
reflect recent patterns. The rates that
follow were adjusted to the 1970 US stan-
dard million population. The incidence rate
of primary CNS malignancies was 27.2 per
million children younger than 20 years of
age (if intracranial germ cell malignancies
are included, the rate was 29.1 per million).
Males (30.0 per million) had a 24% higher
incidence rate relative to females (24.2 per
million). Figures III.5 and III.6 illustrate
the sex-specific rates by histologic groups of
children younger than 20 years of age and
younger than 15 years of age, respectively.
A clear male preponderance for both PNET
and ependymomas was evident, but rates
for males and females were similar for the
other histologic groups.
Black-white differences in incidence
White children (28.5 per million) had
an 18% higher average CNS incidence rate
compared with black children (24.2 per
million). Figure III.7 depicts overall inci-
Figure III.5: Malignant CNS tumor age-adjusted*
incidence rates by histologic group and sex
age <20, all races, SEER, 1990-95
30
14.8
7.3
4.5
3
0.5
24.2
13.5
4.2
4.4
1.5
0.6
All CNS
Astrocytomas
PNET
Other gliomas
Ependymomas
Other CNS
0 4 8 12162024283236
Average annual rate per million
Males
Females
*Adjusted to the 1970 US standard population
32.7
15.7
8.6
4.5
3.5
0.5
26.8
14.5
5
5
1.8
0.4
All CNS
Astrocytomas
PNET
Other gliomas
Ependymomas
Other CNS
0 4 8 12162024283236
Average annual rate per million
Males
Females
*Adjusted to the 1970 US standard population
Figure III.6: Malignant CNS tumor age-adjusted*
incidence rates by histologic group and sex
age <15, all races, SEER, 1990-95

ICCC III CNS
56
National Cancer Institute
SEER Pediatric Monograph
dence rates by sex for white children, black
children, and all children combined. It is
evident that the racial difference in CNS
rates was primarily concentrated among
males. There was only a slightly higher
CNS cancer incidence rate among white
compared with black females (8%), while
the racial difference in rates for males was
somewhat more pronounced (26%).
TRENDS
The observation that CNS cancer
incidence in children appears to have
increased in the past two decades has been
the subject of numerous previous reports
[5-8]. There is considerable debate regard-
ing the possible reasons for the apparent
trend. One concern is that changes in
environmental exposures may be respon-
sible for the increasing incidence, although
epidemiologic evidence to support this
hypothesis currently is lacking [9]. An
alternative explanation is that changes in
reporting due to improvements in diagnos-
tic technology and case ascertainment may
be contributing to the increasing trend.
Figure III.8 illustrates the increase in
incidence rates of CNS cancer for the years
1975-95 for children younger than 15 years
of age. Based on a model using a constant
rate of increase in incidence over this
period, the estimated annual percentage
change (EAPC) was +1.5% (continuous
green line in Figure III.8). Smith et al [5]
recently evaluated CNS trends for children
in the United States from SEER data using
a more sophisticated statistical modeling
technique. They demonstrated that the
incidence of CNS malignancies did not
increase steadily from 1973 to 1994, but
rather “jumped” to a steady, but higher rate
after 1984-85. When the same methodol-
ogy was applied to the younger than 15
year old age group described in this chapter
for the years 1975 to 1995, this “jump
model”, with the optimal change point from
lower to higher incidence occurring after
1985, produced a significantly better fit
than the model using a constant linear rate
Figure III.7: Malignant CNS tumor age-adjusted*
incidence rates by race and sex
age <20, all races, SEER, 1990-95
30
31.5
25
24.2
25.3
23.4
All Races
White
Black
0 4 8 1216202428323640
Average annual rate per million
Males
Females
*Adjusted to the 1970 US standard population
Figure III.8: Temporal trends in malignant CNS tumor
age-adjusted* incidence rates, age <15
all races, both sexes, SEER, 1975-95
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
75 77 79 81 83 85 87 89 91 93 95
Year of diagnosis
0
5
10
15
20
25
30
35
40
Average annual rate per million
Incidence
1975-95
1975-85
1986-95
)
*Adjusted to the 1970 US standard population
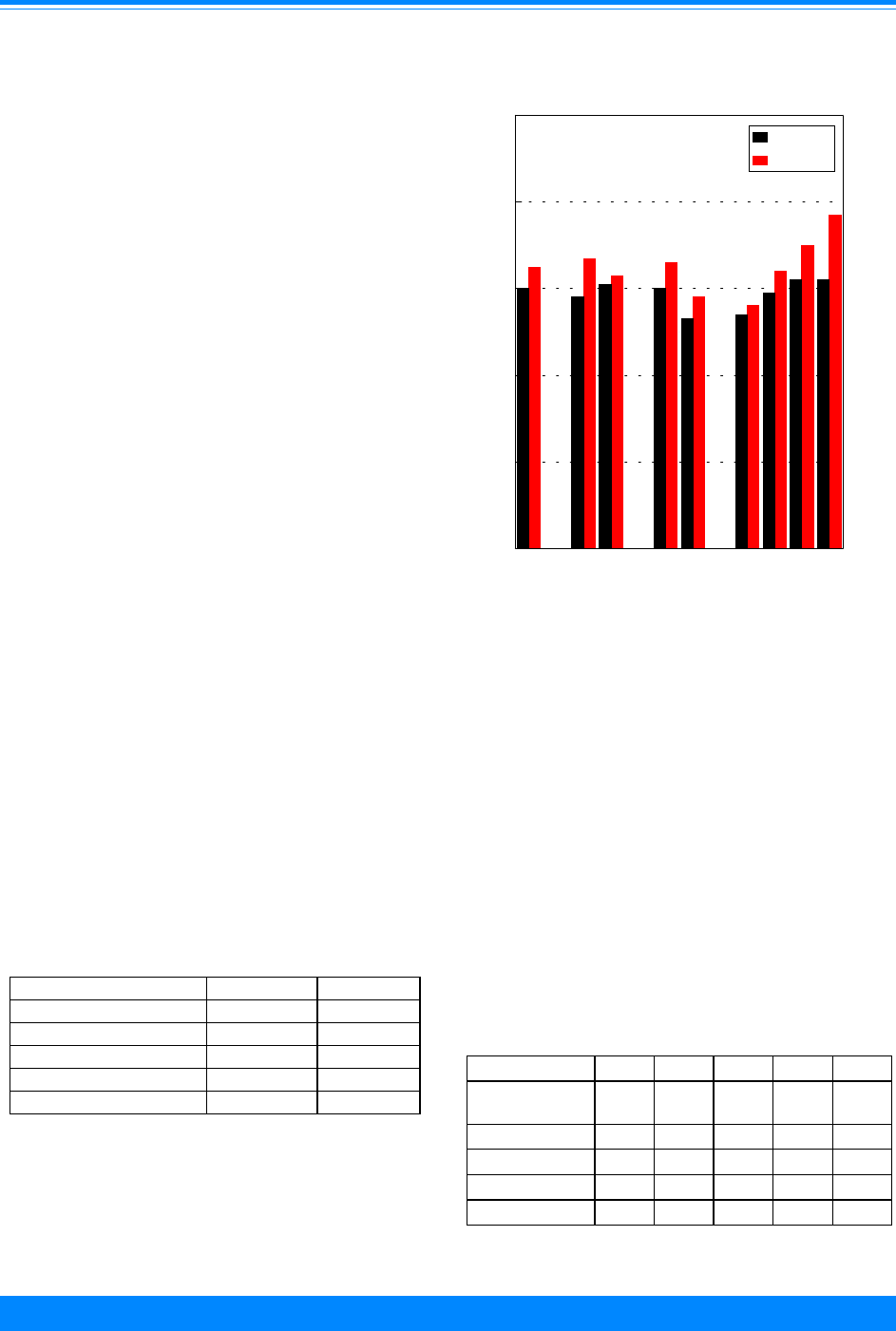
ICCC IIICNS
57
National Cancer Institute SEER Pediatric Monograph
of increase (p = 0.003). The EAPC from
1975-84 was –0.1% (blue line in Figure
III.8) and for 1986-95 the EAPC was also
–0.1% (red line in Figure III.8). The timing
of the jump in incidence is coincident with
the wide-scale availability of magnetic
resonance imaging (MRI) in the United
States [5]. This observation, combined with
the absence of any jump in CNS cancer
mortality during the same period, lends
support to the contention that improved
diagnosis and reporting during the 1980’s is
largely responsible for the temporal trends
in CNS incidence rates that have been
observed since the 1970s. Whether the
relatively stable rates of childhood CNS
cancer observed over the past decade in the
US will continue, however, remains to be
seen.
SURVIVAL
Although survival differs by histology,
behavior, size and location of the malig-
nancy, in general children with CNS cancer
do not share the favorable prognosis of
those with many other common pediatric
neoplasms, such as acute lymphoblastic
leukemia. Additionally, for children who do
survive CNS cancer, long term morbidity
can be substantial. Table III.1 provides 5-
year relative survival probabilities by
histologic group within 2 time periods.
Survival probability improved
somewhat over the two time periods. Nev-
ertheless, other than astrocytomas, many of
which were low grade malignancies such as
Figure III.9: Total malignant CNS tumor 5-year relative
survival rates by sex, race, age and time period
SEER (9 areas), 1975-84 and 1985-94
60
58
61
60
53
54
59
62 62
65
67
63
66
58
56
64
70
77
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex Race
Age
juvenile pilocytic astrocytomas, survival
probability was less than 60%. While there
were only minimal differences in survival of
CNS cancer by sex and race, age was an
important factor. Table III.2 provides 5-
year relative survival for 1986-94 according
to age and histologic groups.
For all CNS cancer combined, survival
probability increased with increasing age.
Very young children with CNS cancer,
especially infants with ependymoma or
PNET, were at particularly high risk of
Table III.2: 5-year relative survival rates for
CNS cancer by type and age group
all races, both sexes, SEER, 1986-94
ICCC Group <1 1-4 5-9 10-14 15-19
All CNS
Cancer
45% 59% 64% 70% 77%
Astrocytoma
69 79 70 75 75
Other Glioma * 51436479
Ependymoma
25 46 71 76 *
PNET
19 46 69 57 75
* less than 20 cases.
Table III.1: 5-year relative survival rates for
CNS by type and time period
age <20, all races, both sexes
SEER 1975-84 and 1985-94
ICCC Group 1975-84 1985-94
All CNS Cancer 60% 65%
Astrocytoma 70 74
Other Glioma 47 57
Ependymoma 39 56
PNET 52 55
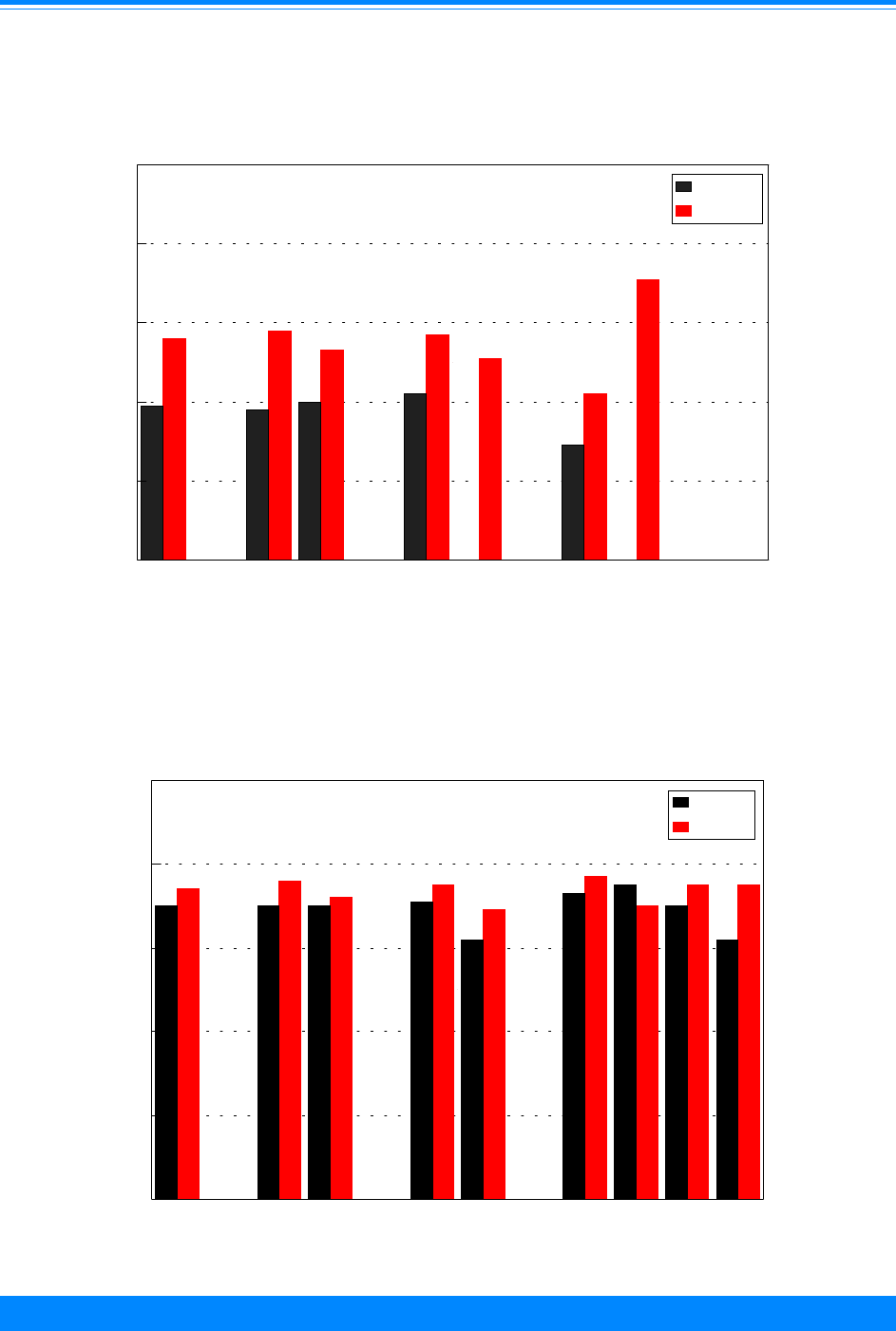
ICCC III CNS
58
National Cancer Institute
SEER Pediatric Monograph
Figure III.11: Astrocytoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas) 1975-84 and 1985-94
70 70 70
71
62
73
75
70
62
74
76
72
75
69
77
70
75 75
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
Figure III.10: Ependymoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
39
38
40
42
29
56
58
53
57
51
42
71
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
# - <25 cases - rate not shown
###
#

ICCC IIICNS
59
National Cancer Institute SEER Pediatric Monograph
Figure III.12: PNET 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
52
46
60
53
52
48 48
57
63
55
58
51
57
54
40
69
57
75
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
Figure III.13: Other gliomas 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
47
49
46
47
49
44
39
48
63
57
61
53
62
41
55
43
64
79
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
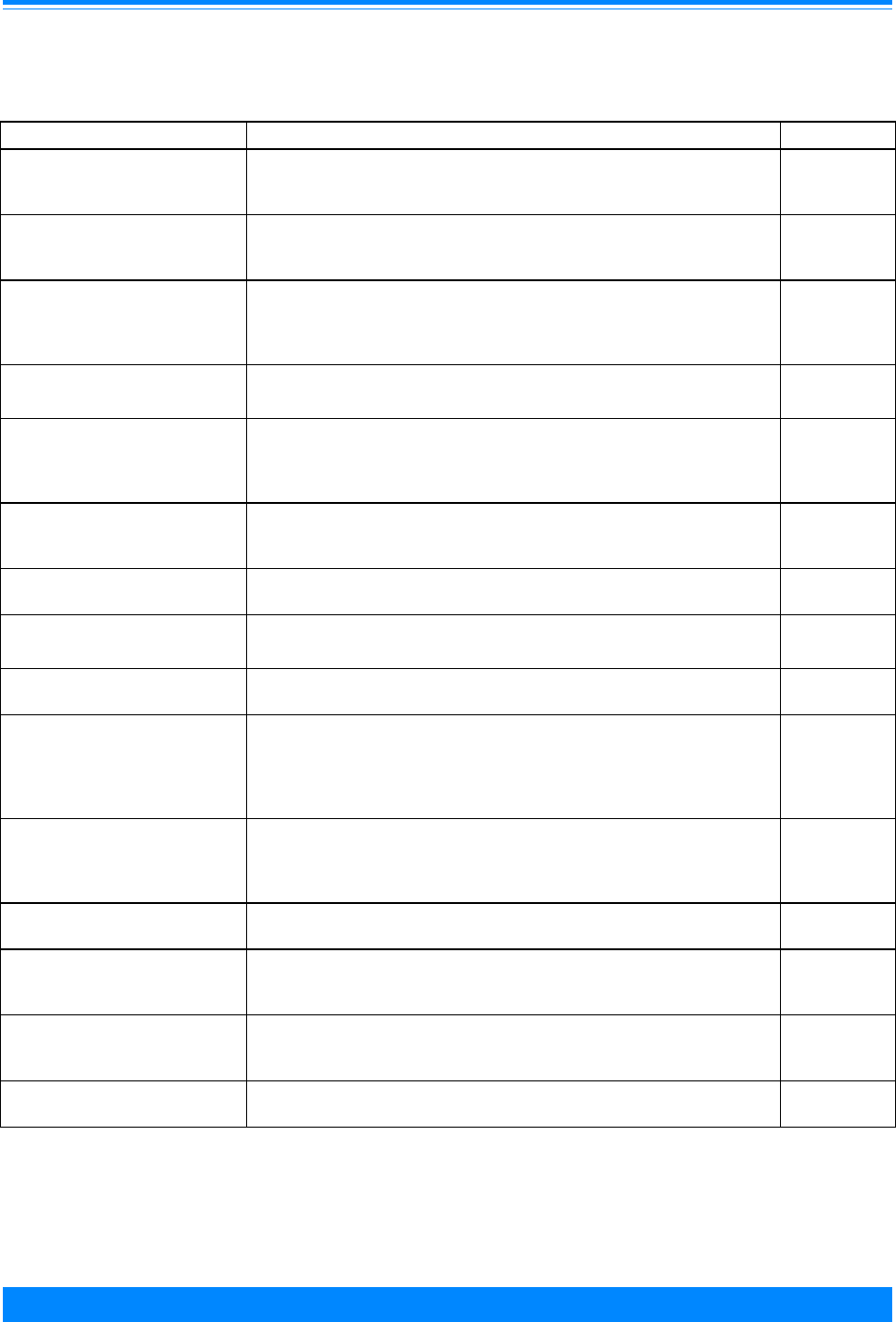
ICCC III CNS
60
National Cancer Institute
SEER Pediatric Monograph
Table III.3: Current knowledge on causes of childhood brain tumors
Exposure or Characteristic Comments References
Sex Incidence of medulloblastoma and ependymomas in males is higher than
in females. For other types of brain tumors, there is little difference
between males and females.
10
Therapeutic doses of ionizing
radiation to head
Children treated for tinea capitis experienced 2.5-6-fold increased risk.
Currently, those at risk are children treated with radiation to the head for
leukemia or a previous brain tumor.
11,12
Neurofibromatosis, tuberous
sclerosis, nevoid basal cell
syndrome, Turcot syndrome, Li-
Fraumeni syndrome
Children with these genetic conditions have a greatly increased risk of
brain tumors, for example, 50-fold for neurofibromatosis and 70-fold for
tuberous sclerosis. Together, these conditions account for less than 5% of
all childhood brain tumors.
10,13,14,28
Maternal diet during pregnancy Frequent cured meat consumption has been consistently associated with a
1.5-2.0 fold increased risk. However, it is unclear whether cured meats or
another dietary factor are responsible, since most aspects of diet have not
yet been studied.
10,13,15-17
Parent or sibling with brain
tumor
Having a sibling or parent with a brain tumor has usually been associated
with a 3-9 fold increased risk. It may be that the excess risk is explained
completely by the specific genetic conditions listed above.
10,13,17,18
Family history of bone cancer,
leukemia or lymphoma.
The increased risk seen in some studies may be explained by the Li-
Fraumeni syndrome.
10,13,22,23,
24
Electromagnetic fields A small increase in risk has been observed in some studies, but not most. 10,13,19,29,
30
Products containing N-nitroso
compounds: beer, incense,
make-up, antihistamines,
diuretics, rubber baby bottle and
pacifier nipples
The data are inconsistent; associations seen in one study have generally
not been reported in later studies.
10,13,21
Father’s occupation and related
exposures
Many associations have been reported, but few have been replicated:
aircraft industry, agriculture, electronics mfg., petroleum industry,
painter, paper or pulp mill worker, printer, metal-related occupation,
exposure to paint, ionizing radiation, solvents, electromagnetic fields.
10,13,25
Pesticides There has been little focused research on this topic. Two small studies
suggest an association with use of no-pest strips.
10,13,20,31
History of head injury This is difficult to study because of the rarity of serious head injury and
the possibility that mothers of children with brain tumors are more likely
than control mothers to recall minor head injuries.
10,13,26
Family history of epilepsy or
seizures
The data are inconsistent. One study suggests that the effect of family
history of seizures may differ by type of brain tumor and/or type and
circumstances of seizures.
13,18,27
Family history of mental
retardation
Increased risk observed in one study of adults and one of children. 13
Note that the majority of these risk factors have been reviewed recently in references 10 and 13; only selected
references are presented for additional reading.
Factors for which evidence
is suggestive but not conclusive
Known risk factors
Factors for which evidence
is inconsistent or limited

ICCC IIICNS
61
National Cancer Institute SEER Pediatric Monograph
mortality. Relative to younger children,
adolescents with CNS cancer tended to fare
well (Figures III.9-III.13).
RISK FACTORS
Table III.3 presents a general sum-
mary of the current knowledge on causes of
brain cancer in children. To date, there is
no specific risk factor known to explain a
substantial proportion of brain tumor
occurrence. Some hereditary conditions
that are clearly associated with increased
susceptibility to CNS cancer in children
include neurofibromatosis type 1, nevoid
basal cell syndrome, and tuberous sclerosis.
These diseases are rare, however, and not
all children with genetic predispositions go
on to acquire cancer. Although a somewhat
increased risk has been observed when a
sibling or parent has had a brain tumor, the
association with family history is not strong
or consistent. Thus, from a population
perspective, known inherited genetic factors
explain only a small percentage of child-
hood CNS cancer incidence. The same can
be said for many other exposures that have
been studied. While therapeutic doses of
ionizing radiation to the head are defini-
tively known to increase the risk of brain
tumors in children, this exposure is largely
historical in nature because therapeutic
head x-rays are now used very sparingly
and with much greater caution than in the
past. There is some evidence that certain
dietary components during pregnancy may
either raise or lower risk, but the relevant
aspects have not yet been clarified. For
exposures with inconsistent or limited data
that are listed in the table, it is not yet
possible to say whether they influence risk.
We know a few factors that do not appear
to increase a child’s risk of developing a
brain tumor, including passive cigarette
smoke exposure, electric blanket use, and
ultrasound testing during pregnancy. The
difficulty in identifying CNS cancer risk
factors may stem in part from studying all
childhood brain tumors as a single entity
when many different histologic subtypes
occur. The rarity of any specific histologic
type makes it very difficult to accrue
enough cases for epidemiologic study.
SUMMARY
Cancer of the brain and central ner-
vous system comprises nearly 17% of
malignancies in children younger than 20
years of age. As a group, CNS cancer is the
most common solid tumor and the second
most common malignancy of childhood.
The overall annual incidence in the United
States is about 27 per million children
younger than 20 years of age (29 per mil-
lion with intracranial germ cell malignan-
cies included). The incidence of CNS cancer
is higher in children younger than 8 years
of age than in older children or adolescents.
This difference is largely attributable to
cerebellar PNET (medulloblastoma), brain
stem gliomas and ependymomas, which all
occur primarily before the age of 10 years.
CNS cancer incidence is slightly higher in
males than in females, largely due to the
male predominance of PNET and ependy-
momas. Rates are higher in white children
than in black children, although the differ-
ences are seen primarily in males and in
young children. Survival, which is depen-
dent on the type and location of the CNS
malignancy, tends to be worse in very
young children than in older children. CNS
cancer incidence rates remained essentially
stable from 1986-95. Unfortunately, the
causes of CNS cancer remain largely unde-
termined. The few definitive risk factors
that are known explain only a small propor-
tion of the total case population.
Reference List
1. Heideman RL, Packer RJ, Albright LA, Free-
man CR, Rorke LB; Tumors of the central
nervous system. Pizzo P, Poplack D, Editors.
Principles and Practices of Pediatric Oncology.
3rd ed. Philadelphia, PA: Lippincott-Raven;
1997:633-697.
2. Kleihues P, Burger PC, Scheithauer BW. The

ICCC III CNS
62
National Cancer Institute
SEER Pediatric Monograph
new WHO classification of brain tumours.
Brain Path. 1993;3:255-268.
3. Kramarova E, Stiller CA. The international
classification of childhood cancer. Int J Cancer
1996;68:759-765.
4. Gurney JG, Wall DA, Jukich PJ, Davis FG. The
contribution of nonmalignant tumors to CNS
tumor incidence rates among children in the
United States. Cancer Causes Control
1999;10:101-105.
5. Smith MA, Feidlin B, Ries LAG, Simon R.
Trends in reported incidence of primary
maligant brain tumors in children in the
United States. J Natl Cancer Inst
1998;90:1269-1277.
6. Gurney JG, Ross JA, Wall DA, Bleyer WA,
Severson RK, Robison LL. Infant cancer in the
U.S.: histology-specific incidence and trends,
1973 to 1992. J Pediat Hematol Oncol
1997;19:428-432.
7. Gurney JG, Davis S, Severson RK, Fang J-Y,
Ross JA, Robison LL. Trends in cancer inci-
dence among children in the U.S. Cancer
1996;78:532-541.
8. Bunin GR, Feuer EJ, Witman PA, Meadows AT.
Increasing incidence of childhood cancer:
report of 20 years experience from the greater
Delaware Valley Pediatric Tumor Registry.
Paediatr Perinat Epidemiol 1996;10:319-338.
9. EPA Conference on Preventable Causes of
Cancer in Children (Minutes). Office of
Children’s Health Protection, U.S. Environ-
mental Protection Agency. Washington, DC
20460, 1997.
10. Preston-Martin S and Mack WJ. Neoplasms of
the nervous system. In: Cancer Epidemiology
and Prevention. D Schottenfeld and JF
Fraumeni eds. Oxford University Press, New
York, 1996;1231-1281.
11. Ron E, Modan B, Boice JD, Alfandary E,
Stovall M, Chetrit A, and Katz L. Tumors of the
brain and nervous system after radiotherapy in
childhood. N Engl J Med 1988;319:1033-1039.
12. Shore RE, Albert RE, and Pasternack BR.
Follow up study of patients treated by X-ray
epilation for tinea capitis: Resurvey of
post-treatment illness and mortality experi-
ence. Arch Environ Health 1976; 31:21-28.
13. Kuijten RR and Bunin GR. Risk factors for
childhood brain tumor: A review. Cancer
Epidemiol Biomarkers Prev 1993;2:277-288.
14. Narod SA, Stiller C, Lenoir GM. An estimate
of the heritable fraction of childhood cancer. Br
J Cancer 1991; 63:993-9.
15. Bunin GR, Kuijten RR, Buckley JD, Rorke LB,
and Meadows AT. Relation between maternal
diet and subsequent primitive neuroectodermal
brain tumors in young children. N Engl J Med
1993; 329:536-541.
16. Bunin GR, Kuijten RR, Boesel CP, Buckley JD,
and Meadows AT. Maternal diet and risk of
astrocytic glioma in children: a report from the
Children’s Cancer Group. Cancer Causes
Control 1994;5:177-187.
17. Preston-Martin S, Pogoda JM, Mueller BA,
Holly EA, Lijinsky W, and Davis RL. Maternal
consumption of cured meats and vitamins in
relation to pediatric brain tumors. Cancer
Epidemiol Biomarkers Prev 1996;5:599-605.
18. Kuijten RR, Strom SS, Rorke LB, Boesel CP,
Buckley JD, Meadows AT, and Bunin GR.
Family history of cancer and seizures in young
children with brain tumors: A report from the
Children’s Cancer Group (United States and
Canada) Cancer Causes and Control
1993;4:455-464.
19. Gurney JG, Mueller BA, Davis S, and Schwartz
SM. Childhood brain tumor occurrence in
relation to residential power line configura-
tions, electric heating sources, and electric
appliance use. Am J Epidemiol 1996;143:120-
128.
20. Davis JR, Brownson RC, Garcia R, Bentz BJ,
and Turner A. Family pesticide use and
childhood brain cancer. Arch Environ Contam
Toxicol 1993;24:87-92.
21. Bunin GR, Buckley JD, Boesel CP, Rorke LB,
and Meadows AT. Risk factors for astrocytic
glioma and primitive neuroectodermal tumor
of brain in young children: A report from the
Children’s Cancer Group. Cancer Epidemiol
Biomarkers Prev 1994;3:197-204.
22. Draper GJ, Heaf MM, and Kinnier Wilson LM.
Occurrence of childhood cancers among sibs
and estimation of familial risks. J Med Genet
1977;14:81-90.
23. Miller RW. Deaths from childhood cancer in
sibs. N Engl J Med 1968;279:122-126.
24. Farwell J and Flannery JT. Cancer in relatives
of children with central-nervous-system
neoplasms. N Engl J Med 1984;311:749-753.
25. McKean Cowdin R, Preston-Martin S, Pogoda
JM, Holly EA, Mueller BA, Davis RL. Parental
occupation and childhood brain tumors –
astroglial and primitive neuroectodermal
tumors. J Occup Environ Med 1998;40:332-
340.
26. Gurney JG, Preston-Martin S, McDaniel AM,
Mueller BA, Holly EA. Head injury as a risk
factor for brain tumors in children: results
from a multicenter case-control study. Epide-
miology 1996;7:485-9.
27. Gurney JG, Mueller BA, Preston-Martin S,
McDaniel AM, Holly EA, Pogoda JM, Davis RL.
A study of pediatric brain tumors and their
association with epilepsy and anticonvulsant
use. Neuroepidemiology 1997;16:248-55.
28. McLendon RE, Tien RD. Genetic syndromes
associated with tumors and/or hamartomas. In
Bigner DD, McLendon RE, Bruner JM: Russell

ICCC IIICNS
63
National Cancer Institute SEER Pediatric Monograph
and Rubinstein’s Pathology of Tumors of the
Nervous System, 6
th
Edition. Arnold; London
GB, 1998: 371-417.
29. Preston-Martin S, Gurney JG, Pogoda JM,
Holly EA, Mueller BA. Brain tumor risk in
children in relation to use of electric blankets
and water bed heaters. Am J Epidemiol
1996;143:1116-1122.
30. Preston-Martin S, Navidi W, Thomas D, Lee P,
Bowman J, Pogoda J. Los Angeles study of
residential magnetic fields and childhood brain
tumors. Am J Epidemiol 1996;143:105-119.
31. Pogoda JM, Preston-Martin S. Household
pesticides and risk of pediatric brain tumors.
Environ Health Perspect 1997; 105:1214-1220.

64
National Cancer Institute
SEER Pediatric Monograph

ICCC IVSYMPATHETIC NERVOUS SYSTEM TUMORS
65
National Cancer Institute
SEER Pediatric Monograph
Marc T. Goodman, James G. Gurney, Malcolm A. Smith, Andrew F. Olshan
HIGHLIGHTS
Incidence
♦ In the US, approximately 700 children and adolescents younger than 20 years of age
are diagnosed with tumors of the sympathetic nervous system each year, of which
approximately 650 are neuroblastomas.
♦ Sympathetic nervous system tumors accounted for 7.8% of all cancers among
children younger than 15 years of age.
♦ Over 97% of sympathetic nervous system tumors are neuroblastomas, embryonal
malignancies of the sympathetic nervous system that occur almost exclusively in
infants and very young children.
♦ Regardless of age, neuroblastomas most commonly occurred in the adrenal gland.
Mediastinal tumors were more frequent in infants than in older children, while the
opposite age pattern was observed for CNS tumors (Figure IV.1).
♦ The average age-adjusted annual incidence rate for all sympathetic nervous system
cancers was 9.5 per million children.
♦ The occurrence of sympathetic nervous system malignancies was strongly
age-dependent (Figure IV.2). For neuroblastomas alone, the incidence rate for both
sexes combined during the second year of life (29 per million) was less than half that
of infancy (64 per million).
♦ Neuroblastomas were by far the most common cancer of infancy, with an incidence
rate almost double that of leukemia, the next most common malignancy that
occurred during the first year of life.
♦ Sixteen percent of infant neuroblastomas were diagnosed during the first month
following birth and 41% were diagnosed during the first 3 months of life (Figure
IV.3).
♦ Over the 21-year observation period, there was little indication of an increase in the
overall incidence of sympathetic nervous system malignancies (Figure IV.4). The
estimated annual percent change in age-adjusted incidence rates was 0.4%.
Survival
♦ For children aged 1 to 4 years at diagnosis, 5-year survival rate improved from 35%
during 1975-84 to 55% during 1985-94. Survival at 5 years from diagnosis was
essentially unchanged over these time intervals among infants (83%) and children 5
years or older (40%).
Risk factors
♦ Relatively little is known about the etiology of sympathetic nervous system tumors
(Table IV.3). The young age at onset of most cases illustrates the need to investigate
exposure events occurring before conception and during gestation.
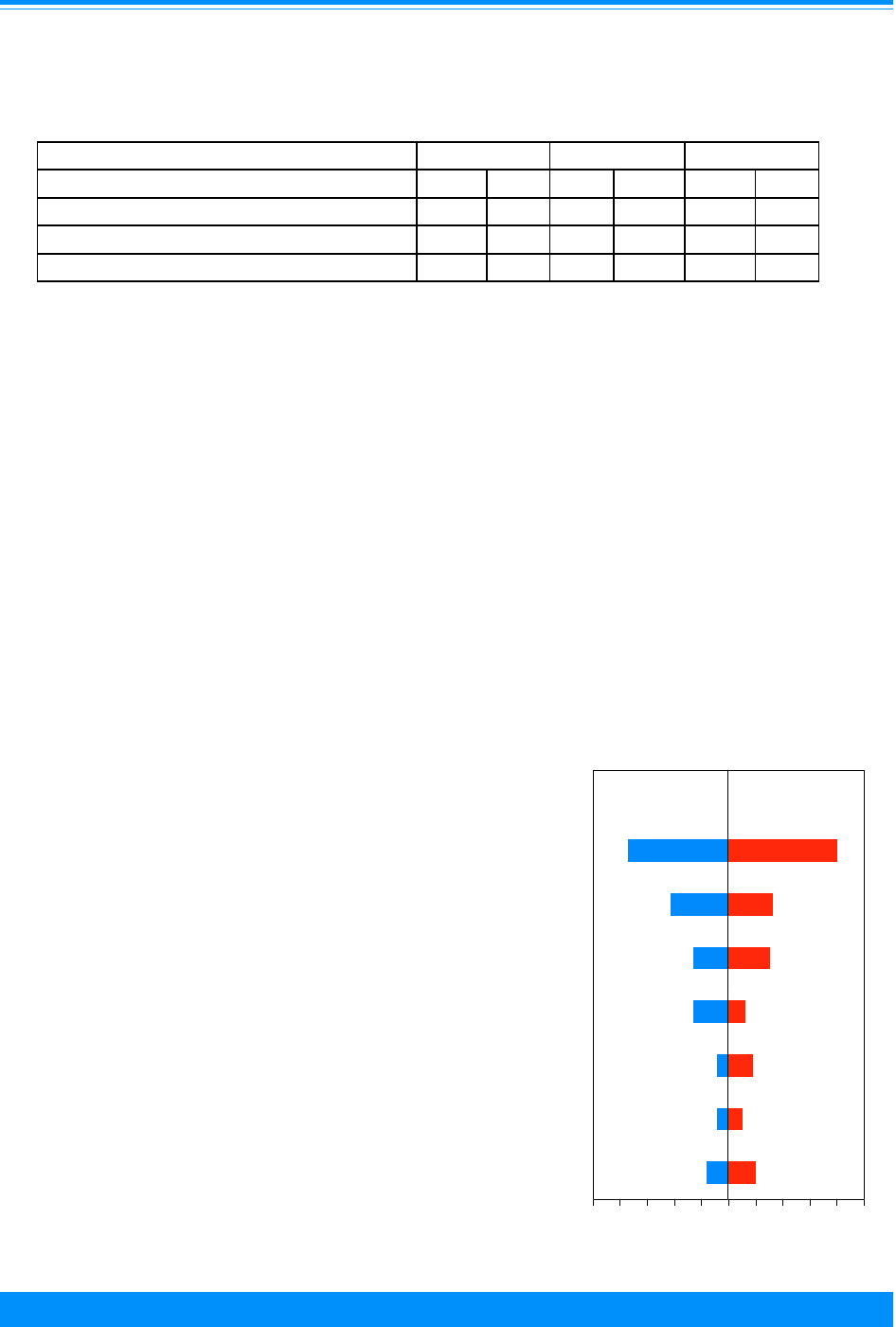
ICCC IV SYMPATHETIC NERVOUS SYSTEM TUMORS
66
National Cancer Institute
SEER Pediatric Monograph
INTRODUCTION
Neuroblastoma is an embryonal malig-
nancy of the sympathetic nervous system
that is derived from primordial neural crest
cells and occurs almost exclusively in
infants and young children [1]. Other
childhood malignancies of the sympathetic
nervous system include
ganglioneuroblastoma, which is a more
differentiated variant of neuroblastoma,
and the histogenetically related pheochro-
mocytoma [2]. Malignant paragangliomas,
medulloepitheliomas, neuroepitheliomas
and olfactory neurogenic tumors are also
cancers of the sympathetic nervous system,
although they are extremely rare in chil-
dren and will not be emphasized. To follow
the convention of the International Classifi-
cation of Childhood Cancer system [3], data
for neuroblastoma and ganglioblastoma are
grouped together as one category (hence-
forth called neuroblastomas), and all other
sympathetic nervous system malignancies
as a second category. Because of important
distinctions in biological characteristics and
prognosis of neuroblastomas in infants (less
than 1 year at diagnosis) compared with
older children (older than 1 year of age at
diagnosis) [1], data are provided to high-
light the epidemiology of both age groups
individually. Additionally, because the
occurrence of neuroblastomas and other
sympathetic nervous system malignancies
are so rare in adolescents, the rate calcula-
tions and discussion are limited to children
younger than 15 years of age. In the US,
approximately 700 children and adoles-
cents younger than 20 years of age are
diagnosed with tumors of the sympathetic
nervous system each year, of which approxi-
mately 650 are neuroblastomas.
INCIDENCE
During the 21-year period from 1975
through 1995, 1,542 children were diag-
nosed with sympathetic nervous system
malignancies in the SEER areas (Table
IV.1). This represented 7.8% of all cancer
Figure IV.1 Percent distribution of neuroblastomas
by primary site and age, all races, both sexes
SEER, 1975-95
37
21
13
13
4
4
8
40
16
15
6
9
5
10
Adrenal gland
Connective,
Retroperitonium
Mediastinum
Central
Autonomic
Other sites
01020304050
Relative percent
0 1020304050
subcutaneous,
soft tissue
nervous
system
nervous
system
Age <1 year
at diagnosis
Age 1+ year
at diagnosis
Table IV.1: Number of cases and age-adjusted* incidence rates per million
by ICCC categories of sympathetic nervous system malignancies
and sex, age <15, all races, SEER, 1975-95
Males Females Total
Tumor Type No. Rate No. Rate No. Rate
Neuroblastomas 787 9.4 705 8.9 1492 9.1
Other sympathetic nervous system 28 0.4 22 0.3 50 0.3
Total 815 9.8 727 9.2 1542 9.5
*Adjusted to the 1970 US standard population
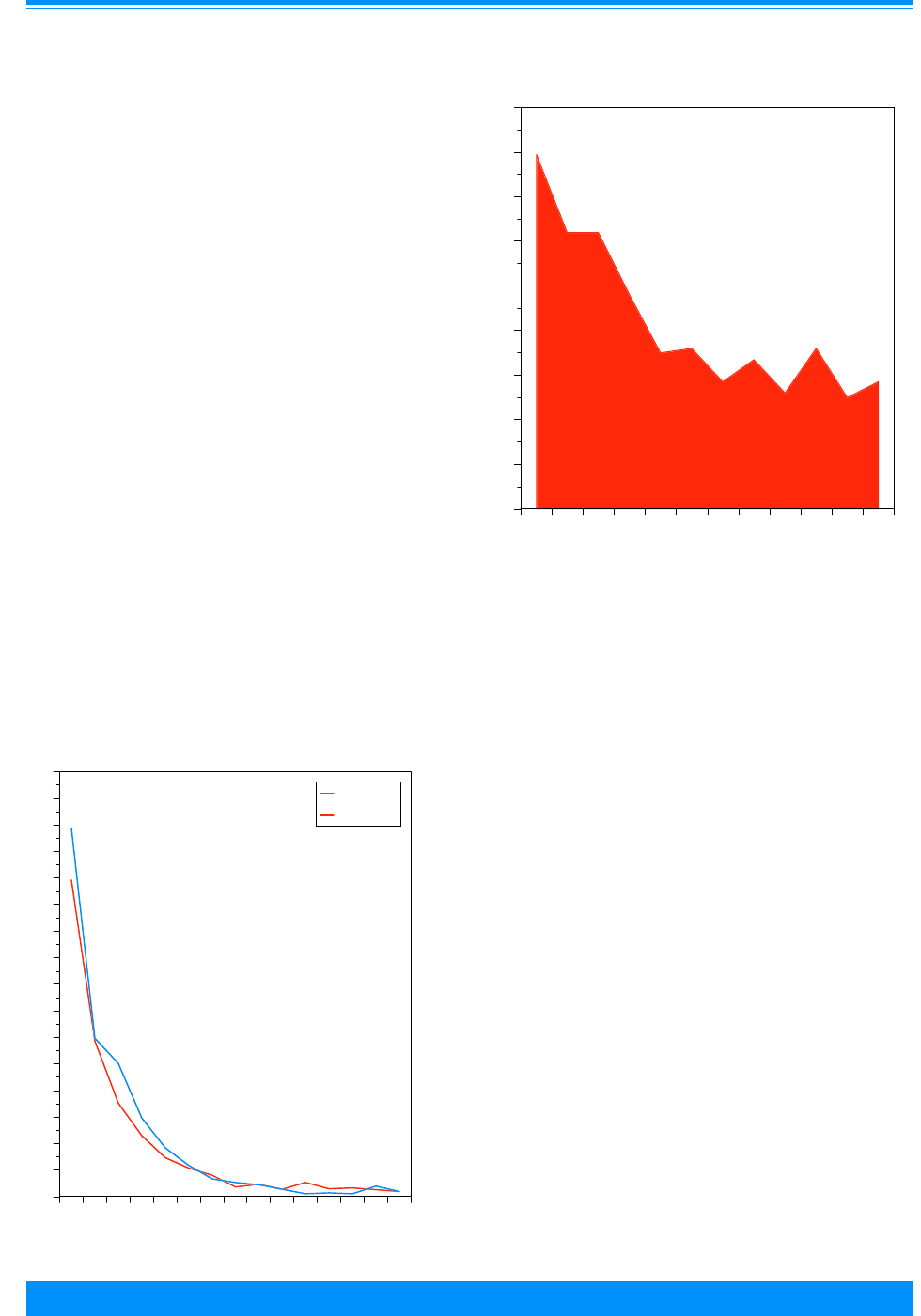
ICCC IVSYMPATHETIC NERVOUS SYSTEM TUMORS
National Cancer Institute SEER Pediatric Monograph
67
in this age group. The majority (97%) of
these malignancies were neuroblastomas;
only 50 children were diagnosed with any
other histological type. Within the neuro-
blastoma category, ganglioneuroblastomas
comprised 15% of tumors (8% among in-
fants and 20% among those 1-14 years of
age).
The distribution of neuroblastomas by
primary site is shown in Figure IV.1. Re-
gardless of age, neuroblastomas most
commonly occurred in the adrenal gland.
Mediastinal tumors were more frequent in
infants than in older children, while the
opposite age pattern was observed for CNS
tumors.
Age-specific incidence
The incidence rate for all sympathetic
nervous system cancers was 9.5 per million
children. The occurrence of sympathetic
nervous system malignancies, however, was
strongly age-dependent. Figure IV.2 illus-
trates the incidence rates by single year of
age
1
and sex, and shows the predominance
of neuroblastomas during infancy. For
neuroblastomas alone, the incidence rate
for both sexes combined during the second
year of life (29 per million) was less than
half that of infancy (64 per million). The
rates for sympathetic nervous system
tumors other than neuroblastomas were
1.2 per million for infants, and less than 1
per million for all other single years of age.
Neuroblastomas were by far the most
common cancer of infancy with an incidence
rate almost double that of leukemia, the
next most common malignancy that occurs
during the first year of life [4]. As shown in
Figure IV.3, 16% of infant neuroblastomas
were diagnosed during the first month
following birth and 41% were diagnosed
during the first 3 months of life.
Figure IV.2: Sympathetic nervous system age-specific
incidence rates by sex, all races
SEER, 1976-84 and 1986-94
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
0123456789101112131415
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
Average annual rate per million
Males
Females
"
)
Figure IV.3: Percent distribution of infant neuroblastomas
by month of age, all races, both sexes, SEER, 1975-95
0123456789101112
Age (in months) at diagnosis
0
2
4
6
8
10
12
14
16
18
Percentage of cases
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.

ICCC IV SYMPATHETIC NERVOUS SYSTEM TUMORS
68
National Cancer Institute
SEER Pediatric Monograph
Sex and race-specific incidence
Figure IV.2 also demonstrates that the
incidence of sympathetic nervous system
cancer was slightly higher among males
than females. For neuroblastomas, overall
male rates (9.8 per million) were 6.5%
higher than female rates (9.2 per million)
with the greatest difference occurring
during infancy (69.3 per million versus 59.6
per million for males and females, respec-
tively). There was no discernable sex
difference for sympathetic nervous system
malignancies other than neuroblastomas.
White infants of both sexes had a
higher incidence of sympathetic nervous
system tumors than did black infants, but
little difference by race was observed
among older children (Table IV.2). The
ratio of white to black incidence rates
among infants was 1.7:1 for males and
1:9:1
for females. In Table IV.2, “all races”
includes whites, blacks, and children of
other identified racial or ethnic back-
grounds. There were too few cases of
sympathetic nervous system among any
other races to calculate reliable incidence
rates.
TRENDS
Over the 21-year observation period,
there was little indication of a linear trend
in the overall incidence of sympathetic
nervous system malignancies (Figure IV.4).
The estimated annual percent change in
age-adjusted incidence rates was 0.37%
(p > 0.05). Rates, however, have increased
somewhat among infants during recent
years. Figure IV.5 shows incidence rates of
neuroblastomas by year of age at diagnosis
for the periods 1976-84 versus 1986-94.
Among infants, the rate in the earlier time
period was 53 per million compared to 74
per million in the later time period. No
differences in rates between the time
Figure IV.4: Trends in sympathetic nervous system
age-adjusted* incidence rates by year of diagnosis
age <15, all races, both sexes, SEER, 1975-95
(
((
(
(
((
(
(
(
(
(
(
(
(
(
(
(
(
(
(
75 77 79 81 83 85 87 89 91 93 95
Year of diagnosis
0
2
4
6
8
10
12
14
16
18
Average annual rate per million
*Adjusted to the 1970 US standard population
Table IV.2: Average annual age-specific incidence rates per million for all
sympathetic nervous system tumors by age, sex, and race
SEER 1975-95
Males Females
Age (in years) at diagnosis White Black All White Black All
<1 83.6 50.5 69.3 74.1 38.4 59.6
1-14 8.2 7.4 7.3 7.6 6.5 6.5
<15* 10.1 8.8 9.8 9.6 8.6 9.2
* Adjusted to the 1970 US standard population

ICCC IVSYMPATHETIC NERVOUS SYSTEM TUMORS
National Cancer Institute SEER Pediatric Monograph
69
periods occurred for children either 1 or 2
years of age at diagnosis. Thus, it does not
appear that the increase among infants can
be explained by a shift towards earlier age
at diagnosis. The increase among infants,
however may be a result of de facto fetal
and neonatal screening. Mass screening of
infants for neuroblastoma has been evalu-
ated in recent years in Japan, Canada, and
some countries in Europe [5,6]. Although
systematic screening for neuroblastoma is
not conducted in the United States, the
awareness of screening in other countries
and the recent widespread availability of
non-invasive diagnostic tests for neuroblas-
toma may have resulted in US physicians
diagnosing cases of neuroblastoma with
minimal clinical symptomatology that
previously were undetected. The docu-
mented ability of some fetal and infant
neuroblastomas to spontaneously regress is
consistent with the hypothesis that the
increased incidence among infants is the
result of detection of cases that were previ-
ously not diagnosed [1,9,10]. Also consis-
tent with this hypothesis is the recent
widespread use of prenatal ultrasound
testing with coincidental detection of adre-
nal neuroblastomas [7,8].
SURVIVAL
Prognosis for neuroblastomas is depen-
dent on age, stage of disease, and the
molecular biologic and cytogenetic charac-
teristics of the tumor [1]. Figure IV.6
illustrates the more favorable prognosis for
infants with neuroblastoma (5-year relative
survival rate, 83%) compared to children
older than 1 year of age. The favorable
outcome for infants with neuroblastoma no
doubt reflects the favorable biological
Figure IV.5: Neuroblastoma age-specifice incidence
rates by age, all races, both sexes
SEER, 1976-84 and 1986-94
53.7
29.3
20.9
9.9 9.9
73.4
28.5
20.7
15.6
6.8
<11234
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
Average annual rate per million
1976-84
1986-94
Figure IV.6: Neuroblastoma 5-year relative survival
rates by sex, race, age, and time period
SEER (9 areas), 1975-84 and 1985-94
54
49
58
55
44
83
35
43
64
62
67
64
61
83
55
40
Total Male Female White Black <1 1-4 5-9
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age

ICCC IV SYMPATHETIC NERVOUS SYSTEM TUMORS
70
National Cancer Institute
SEER Pediatric Monograph
characteristics of neuroblastomas arising in
this age group [1]. For children aged 1 to 4
years at diagnosis, the 5-year survival rate
improved from 35% during 1975-84 to 55%
during 1985-94. Survival was essentially
unchanged during these time intervals for
children older than 4 years of age (40%).
There were no substantive differences in
survival by sex or race (Figure IV.6).
RISK FACTORS
Relatively little is known about the
etiology of sympathetic nervous system
tumors (Table IV.3). The young age at
onset of most cases illustrates the need to
investigate exposure events occurring
before conception and during gestation.
The few epidemiological investigations of
Table IV.3: Current knowledge on causes of neuroblastoma (NB)
Exposure or Characteristic Comments References
Medications Two studies have reported increased risk when mothers
took medications during pregnancy such as
amphetamines, diuretics, tranquilizers, or muscle
relaxers or for vaginal infection. Other studies have
reported an association with maternal phenytoin
treatment.
11,12,13
Hormones Two studies reported that sex hormones were associated
with an increase in risk. One of the studies reported a 10-
fold increased risk for fertility drug use prior to
pregnancy.
12,13,14
Birth characteristics One study reported increased risk associated with low
birth weight and protective effect for preterm delivery.
This was not confirmed in two other studies.
13,15,16
Congenital anomalies A variety of congenital anomalies has been reported to
occur with NB in a small number of cases, but no
consistent pattern of association has been shown.
11
Previous spontaneous
abortion/fetal death
Previous spontaneous abortion was associated with
increased risk in one study and decreased risk in another.
13,16
Alcohol One study reported a dose-response relationship between
frequency of alcohol use during pregnancy and NB, but
another reported no effect. An association with fetal
alcohol syndrome has also been reported.
12,13,17
Tobacco An early study reported no effect of maternal smoking on
risk. However, a later study suggested a weak dose-
response relationship between level of maternal smoking
during pregnancy and NB risk.
12,13
Paternal occupational exposures Three studies have reported conflicting results on the risk
associated with paternal employment in electronics,
agriculture, and packaging and materials handling.
Specific associated occupational exposures include
electromagnetic fields, pesticides, hydrocarbons, dusts,
rubber, paint, and radiation.
18-20
Factors for which evidence is
inconsistent or limited

ICCC IVSYMPATHETIC NERVOUS SYSTEM TUMORS
National Cancer Institute SEER Pediatric Monograph
71
neuroblastoma have not had sufficient
statistical power or detailed data collection
to provide convincing evidence of etiologic
risk factors. Medications [11,12,13] and
hormones used during pregnancy [12,13,14]
are among the most suggestive factors
suspected to increase the risk of neuroblas-
toma. Certain birth characteristics, pesti-
cide exposure, and parental occupational
exposure to electromagnetic fields
[13,15,16,18-20] have been evaluated, but
with conflicting results. In addition, clinical
and molecular characteristics, such as
amplification of the nmyc oncogene, loss of
heterozygosity of the short arm of chromo-
some 1, and hyperdiploidy, may be useful in
establishing homogenous disease sub-
groups for future epidemiological investiga-
tions of neuroblastoma [1].
SUMMARY
Sympathetic nervous system malignancies,
of which neuroblastomas comprised 97% of
the total, represented 7.8% of cancer in
children younger than 15 years of age. The
incidence rate was 9.5 per million children,
however rates were strongly age-depen-
dent. The incidence rate of sympathetic
nervous system malignancies among in-
fants was 65 per million, and the rate
dropped by half in the second year of life.
Overall, incidence rates did not change
substantially during the study period.
Among infants, however, there was an
increase in incidence rates from 1986-94
compared with the period 1976-84. This
increase was not noted in older children,
thus excluding earlier age at diagnosis as a
likely explanation for the trend. Rather,
the increase likely arose from identification
of previously undetected cases with mini-
mal clinical symptomatology through
widespread application of fetal ultrasound
testing and noninvasive diagnostic tests for
neuroblastoma. The known propensity of
the neuroblastomas of infancy to undergo
spontaneous regression supports this
explanation. Five-year relative survival of
neuroblastomas was 83% for infants, 55%
for children 1-4 years of age, and 40% for
older children. Unfortunately, there is very
little known about why neuroblastoma
occurs, or what factors increase risk for
occurrence.
Reference List
1. Brodeur GM, Castleberry RP. Neuroblastoma.
In: Pizzo PA, Poplack DG, Editors. Principles
and Practices of Pediatric Oncology. 3rd ed.
Philadelphia, PA: Lippencott-Raven; 1997:761-
797.
2. Russell DS, Rubinstein LJ. Pathology of
Tumours of the Nervous System, 5
th
edition.
Williams & Wilkins, Baltimore, MD, 1989; 898-
943.
3. Kramarova E, Stiller CA. The international
classification of childhood cancer. Int J Cancer.
1996;68:759-765.
4. Gurney JG, Ross JA Wall DA Bleyer WA,
Severson RK, Robison LL. Infant cancer in the
U.S.: histology-specific incidence and trends,
1973-1992. J Pediatr Hematol Oncol 1997;
19:428-432.
5. Woods WG, Tuchman M, Robson LL, Bernstein
M, Leclerc JM, Brisson LC, Brossard J, Hill G,
Shuster J, Luepker R, Byrne T, Weitzman S,
Bunin G, Lemieux. A population-based study of
the usefulness of screening for neuroblastoma.
Lancet 1996; 348:1682-1687.
6. Parker L, Powell J. Screening for neuroblas-
toma in infants younger than 1 year of age:
review of the first 30 years. Med Pediatr Onc
1998; 31:455-469.
7. Moore RM, Jeng LL, Kaczmarek RG, Placek
PJ. Use of diagnostic imaging procedures and
fetal and monitoring devices in the care of
pregnant women. Public Health Rep 1990;105:
471-475.
8. Archarya S, Jayabose S, Kogan SJ, Tugal O,
Beneck D, Leslie D, Slim M. Prenatally
diagnosed neuroblastoma. Cancer 1997;
80:304-310.
9. Carlsen NLT. Neuroblastoma: epidemiology
and pattern of regression. Problems in inter-
preting results of mass screening. J Pediatr
Hemat Oncol 1992;14:103-110.
10. Evans AE, Chatten J, D’Angio GJ, Gerson JM,
Robinson J, Schnaufer L. A review of 17 IV-S
neuroblastoma patients at the Children’s
Hospital of Philadelphia. Cancer 1980;45:833-9.
11. Bodeur GM. Neuroblastoma and other periph-
eral neuroectodermal tumors. Chapter 24. In:
Clinical Pediatric Oncology, 4
th
Ed. Fernbach
DJ, Vietti TJ (Eds.). Mosby Yearbook, St. Louis,
MO., 1991.

ICCC IV SYMPATHETIC NERVOUS SYSTEM TUMORS
72
National Cancer Institute
SEER Pediatric Monograph
12. Kramer S, Ward E, Meadows AT, and Malone
KE. Medical and drug risk factors associated
with neuroblastoma: A case control study. J
Natl Cancer Inst 1987;78:797-804.
13. Schwartzbaum JA. Influence of mother’s
prenatal drug consumption on risk of neuro-
blastoma in the child. Am J Epidemiol
1992;135:1358-1367.
14. Michalek AM, Buck GM, Nasca PC, Freedman
AN, Baptiste MS, and Mahoney MC. Gravid
health status, medication use, and risk of
neuroblastoma. Am J Epidemiol 1996;143:996-
1001.
15. Johnson CC and Spitz MR. Neuroblastoma: A
case control analysis of birth characteristics. J
Natl Cancer Inst 1985;74:789-192.
16. Neglia JP, Smithson WA, Gunderson P, King
FL, Singher LJ, and Robison LL. Prenatal and
perinatal risk factors for neuroblastoma. A
case-control study. Cancer 1988;61:2202-2206.
17. Kinney H, Faix R, and Brazy J. The fetal
alcohol syndrome and neuroblastoma. Pediat-
rics 1980;66:130-132.
18. Bunin GR, Ward E, Kramer S, Rhee CA, and
Meadows AT. Neuroblastoma and parental
occupation. Am J Epidemiol 1990;131:776-780.
19. Spitz MR and Johnson CC. Neuroblastoma and
paternal occupation. Am J Epidemiol
1985;121:924-929.
20. Wilkins JR and Hundley VD. Paternal occupa-
tional exposure to electromagnetic fields and
neuroblastoma in offspring. Am J Epidemiol
1990;131:995-1008.

ICCC V
73
National Cancer Institute
SEER Pediatric Monograph
RETINOBLASTOMA
HIGHLIGHTS
Incidence
♦ Retinoblastoma accounted for approximately 11% of cancers developing in the first
year of life, but for only 3% of the cancers developing among children younger than
15 years of age.
♦ In the US, approximately 300 children and adolescents younger than 20 years of age
are diagnosed with retionblastomas each year.
♦ The vast majority of cases of retinoblastoma occur among young children, with
almost two-thirds (63%) of all retinoblastomas occurring before the age of two years
and 95% occurring before the age of five years.
♦ The incidence of bilateral tumors was strongly age dependent with 42% of the retino-
blastomas occurring in children less than one year of age being bilateral compared to
21% of those among children aged one year, and only 9% among older children.
♦ Rates of retinoblastoma were essentially equal among males (3.7 per million) and
females (3.8 per million) and among whites (3.7 per million) and blacks (4.0 per
million) (Table V.2)
♦ There was no substantial sustained change in retinoblastoma incidence during the
21-year period, 1975-95 (Figure V.3 and Table V.2).
Survival
♦ Survival for children with retinoblastoma was quite favorable, with more than 93%
alive at five years after diagnosis. Males and females had similar 5-year survival
rates for the period 1976-94 (93-94%). Black children had slightly lower 5-year
survival rates than white children (89% versus 94%) (Figure V.5).
Risk factors
♦ A retinoblastoma gene has been identified. Each child of a parent with familial
bilateral retinoblastoma has a 50% risk of inheriting the retinoblastoma gene. Some
patients develop the gene as the result of a new mutation (sporadic heritable retino-
blastoma) and can pass the gene on to their children even though they did not
inherit the gene from their parents. Children who inherit the retinoblastoma gene
have a 90% risk of developing retinoblastoma. Genetic retinoblastomas are more
likely to be bilateral and to occur during the first year of life. Little is know about
non-genetic (sporadic) retinoblastomas (Table V.3).
INTRODUCTION
Retinoblastoma is a tumor of childhood
which arises in the retina of the eye and
extremely rarely in the pineal gland [1].
Two types of retinoblastomas have been
described: those linked to genetic muta-
tions and the so-called sporadic retinoblas-
tomas. The genetic-linked retinoblastomas
are divided into two groups, those which
arise in children who carry the retinoblas-
toma gene inherited from one or both
parents (familial retinoblastoma) and those
in which the disease occurs as the result of
a new mutation, usually in their father’s
sperm but sometimes in their mother’s egg
(sporadic heritable retinoblastoma) [2,3].
Both familial retinoblastomas and sporadic
John L. Young, Jr., Malcolm A. Smith, Steven D. Roffers, Jonathan M. Liff, Greta R. Bunin
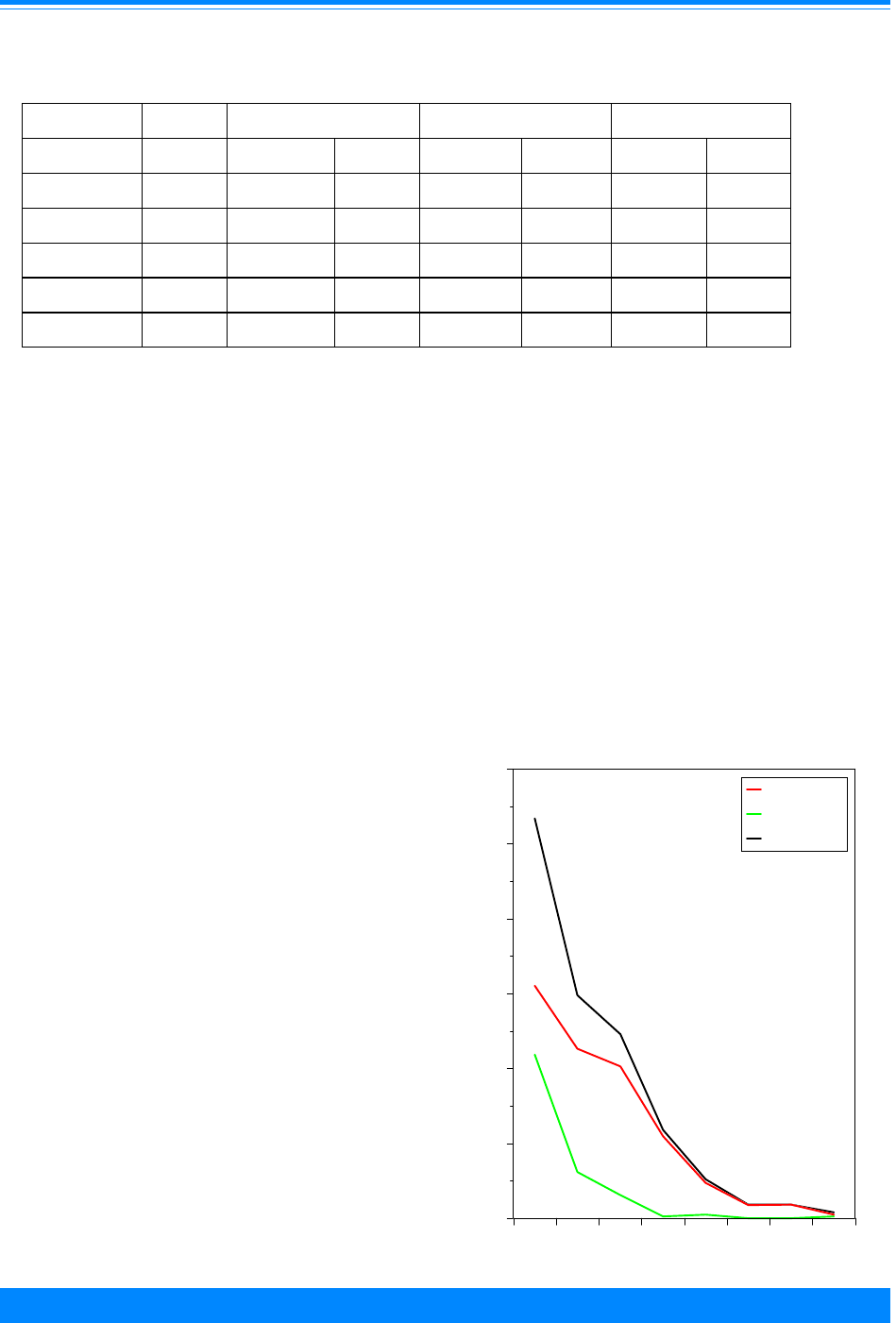
ICCC V RETINOBLASTOMA
74
National Cancer Institute
SEER Pediatric Monograph
heritable retinoblastomas are more likely
to be bilateral and to occur during the first
year of life, while the sporadic retinoblasto-
mas are more likely to be unilateral and
occur after the first year of life.
The importance of retinoblastoma to
cancer research far exceeds the low inci-
dence of this uncommon tumor, since it was
through careful analysis and insightful
mathematical modeling of the age distribu-
tion of unilateral and bilateral cases that
the “tumor suppressor gene” concept was
initially developed [4,5]. Subsequent work
led to the localization of the gene respon-
sible for retinoblastoma to a small region
on the long arm of chromosome 13 [6], and
eventually to isolation of the gene itself [7].
The retinoblastoma gene product is now
recognized as a critical element in control-
ling progression through the cell cycle, and
abnormalities of the retinoblastoma gene
are among the most common occurring in
all types of cancer cells [5,8]. In the US,
approximately 300 children and adoles-
cents younger than 20 years of age are
diagnosed with retionblastomas each year.
INCIDENCE
Table V.1 shows the distribution of
retinoblastomas diagnosed among residents
of the SEER areas during 1975-95 by
laterality, race, and sex. Approximately
one-fourth of all retinoblastomas were
bilateral. All bilateral disease is hereditary
whereas unilateral disease may or may not
be hereditary. The percentage of unilateral
and bilateral tumors were similar for black
children and white children and for males
and females.
The vast majority of cases of retino-
blastoma occur among young children
(Figure V.1), with almost two-thirds (63%)
Table V.1: Number of retinoblastomas by laterality, sex, and race, age <15
SEER, 1975-95
Total Unilateral Bilateral Unknown
No. No. % No. % No. %
Total 625 453 72.5 154 24.6 18 2.9
Males 314 226 72.0 76 24.2 12 3.8
Females 311 227 73.0 78 25.1 6 1.9
Whites 474 341 71.9 116 24.5 17 3.6
Blacks 86 61 70.9 25 29.0 - -
Figure V.1: Unilateral and bilateral retinoblastoma
age-specific incidence rates, all races
both sexes, SEER, 1976-84 and 1986-94
#
#
#
#
#
#
##
(
(
(
(
(
(
((
,
,
,
,
,
,
,
,
012345678
Age (in years) at diagnosis
0
5
10
15
20
25
30
Average annual rate per million
Unilateral
Bilateral
Total
,
(
#

ICCC VRETINOBLASTOMA
75
National Cancer Institute SEER Pediatric Monograph
of all retinoblastomas diagnosed between
1975-95 among children residing in the
SEER areas occurring before the age of two
years and with 95% occurring before the
age of five years. Since retinoblastoma was
extremely rare after the age of five years
(only 28 childhood retinoblastomas re-
ported to SEER areas as having been
diagnosed younger than 20 years of age
occurred in children aged 15-19 years in
comparison to 625 cases in children
younger than 15 years of age), information
presented in this chapter will be limited to
children younger than 15 years of age.
The incidence of bilaterality was
strongly age dependent, with 42% (103/248)
of the retinoblastomas occurring in children
less than one year of age being bilateral
compared to 21% (31/147) of those among
children aged one year, and only 9% (20/
230) among older children. The incidence
rates for both unilateral and bilateral
retinoblastoma decrease as age increases.
The incidence rate for bilateral retinoblas-
toma drops to almost zero after age 2, while
the rate for unilateral retinoblastoma
remains higher until after age 7 (Figure
V.1). Figure V.2 provides a more detailed
view of incidence in the first 3 years of life,
with incidence for the first year of life being
estimated by two month age intervals. For
bilateral tumors, the peak incidence is at 4-
5 months of age, with a sharp decline
thereafter and with very low rates by the
third year of life. For unilateral tumors,
peak incidence is also in the first year of life
(at 6-7 months), but the decline in incidence
with increasing age is much more gradual
than for bilateral tumors, with rates re-
maining above 10 per million children for
the first 3 years of life.
The incidence rate of retinoblastoma
for the period 1975-95 was 3.8 per million
(Table V.2). Retinoblastoma accounted for
approximately 11% of the cancers develop-
ing in the first year of life, but for only 3%
of the cancers developing among children
younger than 15 years of age. Rates of
retinoblastoma among males (3.7 per
million) and females (3.8 per million) were
essentially equal. Rates for whites (3.7 per
million) and blacks (4.0 per million) were
also similar.
Figure V.2: Unilateral and bilateral retinoblastoma
age-specific incidence rates, age <3, all races
both sexes, SEER, 1976-84 and 1986-94
Age (in months) at diagnosis
(
(
(
(
(
(
(
(
,
,
,
,
,
,
,
,
0122436
0
5
10
15
20
25
Average annual rate per million
Unilateral
Bilateral
,
(
< 2
2-3
4-5
6-7
8-9
10-11
Table V.2: Average annual age-adjusted* incidence rates
per million of retinoblastoma, by time period,
race, and sex, age <15 SEER, 1975-95
1975-95 1975-79 1980-84 1985-89 1990-95
All races, Both sexes
3.8 3.6 3.6 3.7 4.0
Male
3.7 3.6 3.3 3.4 4.2
Female
3.8 3.6 4.0 4.0 3.8
White
3.7 3.3 3.5 3.7 4.0
Black
4.0 4.6 4.0 3.7 3.8
* Adjusted to the 1970 US standard population
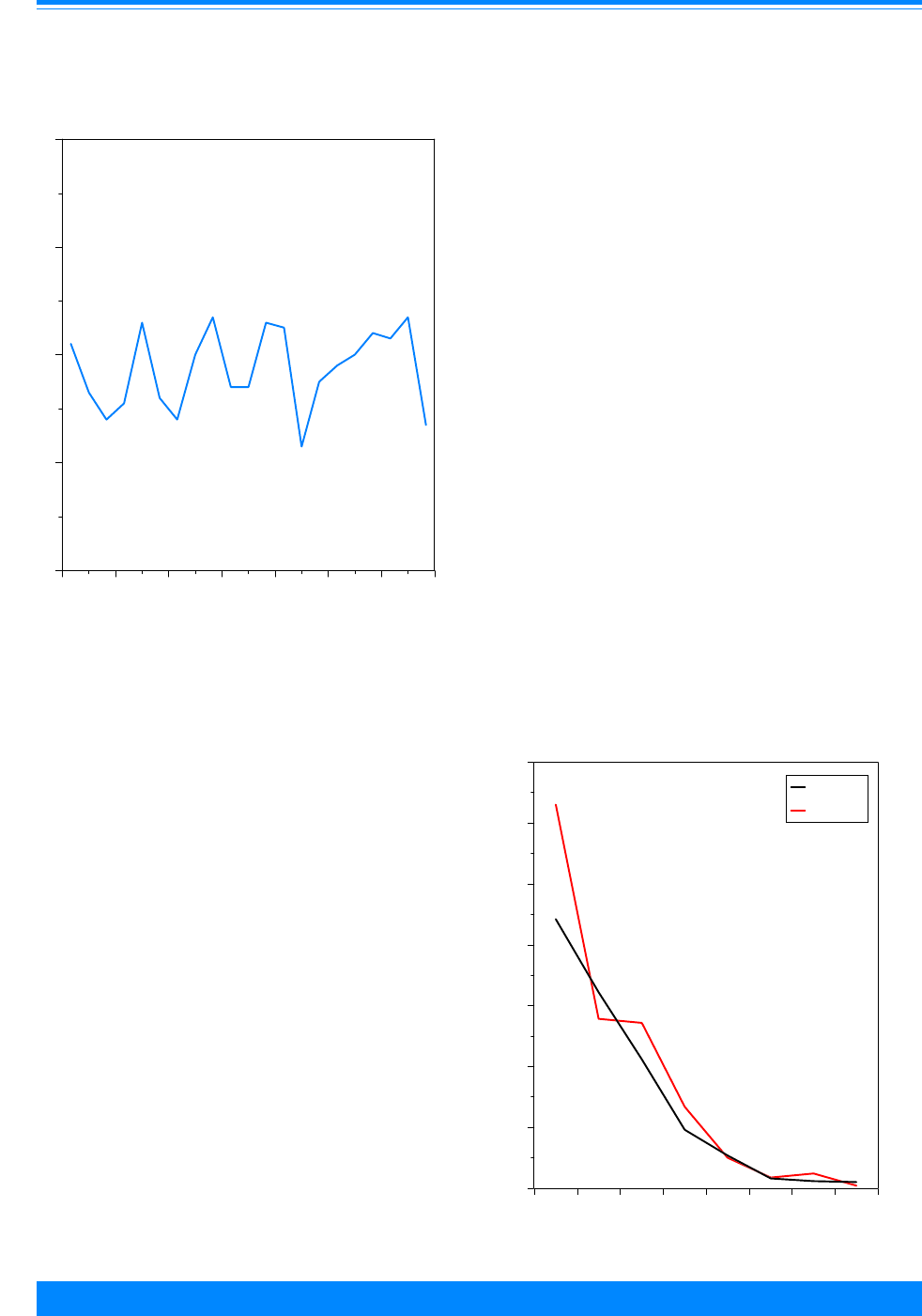
ICCC V RETINOBLASTOMA
76
National Cancer Institute
SEER Pediatric Monograph
TRENDS
Figure V.3 shows the incidence for
retinoblastoma among children younger
than 15 years of age for 1975-95. Because
of the relatively small numbers of children
with retinoblastoma diagnosed in SEER
areas annually (approximately 20 per
year), there was considerable variability in
the year-to-year rates. There was no sub-
stantial sustained change in retinoblas-
toma incidence during this 21 year period.
Table V.2 shows the incidence rate for
retinoblastoma among children younger
than 15 years of age for four specific time
periods between 1975-95 by sex and race.
Rates were slightly higher in the last time
period for males and for whites. The esti-
mated overall annual percent change was
about one-half of one percent per year
(0.6%). The change was greater for males
(1.2%) than for females (0.2%), but neither
change was significant. The rates for
blacks were higher than whites for the
earlier time period but by the late 1980s
were similar. Figure V.4 shows the inci-
dence of retinoblastoma by year of age for
an early time period (1976-84) and for a
recent time period (1986-94) and illustrates
that the small increase that did occur
between these two time periods was prima-
rily the result of increased diagnosis of
retinoblastoma in the first year of life.
SURVIVAL
Figure V.5 shows that survival for
children diagnosed with retinoblastoma in
the period 1975-94 was quite favorable,
with more than 93% alive at five years
after diagnosis. Males and females had
similar 5-year survival rates for the period
1975-94 at 93-94%, while black children
had slightly lower 5-year survival rates
than white children (89% versus 94%).
Figure V.3: Trends in retinoblastoma age-adjusted*
incidence rates, age <15, all races
both sexes, SEER 1975-1995
+
+
+
+
+
+
+
+
+
+
++
+
+
+
+
+
+
+
+
+
1975 1978 1981 1984 1987 1990 1993 1996
Year of diagnosis
0
2
4
6
8
Average annual rate per million
*Adjusted to the 1970 US standard population
Figure V.4: Retinoblastoma age-specific incidence
rates by time period, all races, both sexes
SEER, 1976-84 and 1986-94
,
,
,
,
,
,
,
,
(
(
(
(
(
(
(
(
012345678
Age (in years) at diagnosis
0
5
10
15
20
25
30
35
Average annual rate per million
1976-84
1986-94
(
,
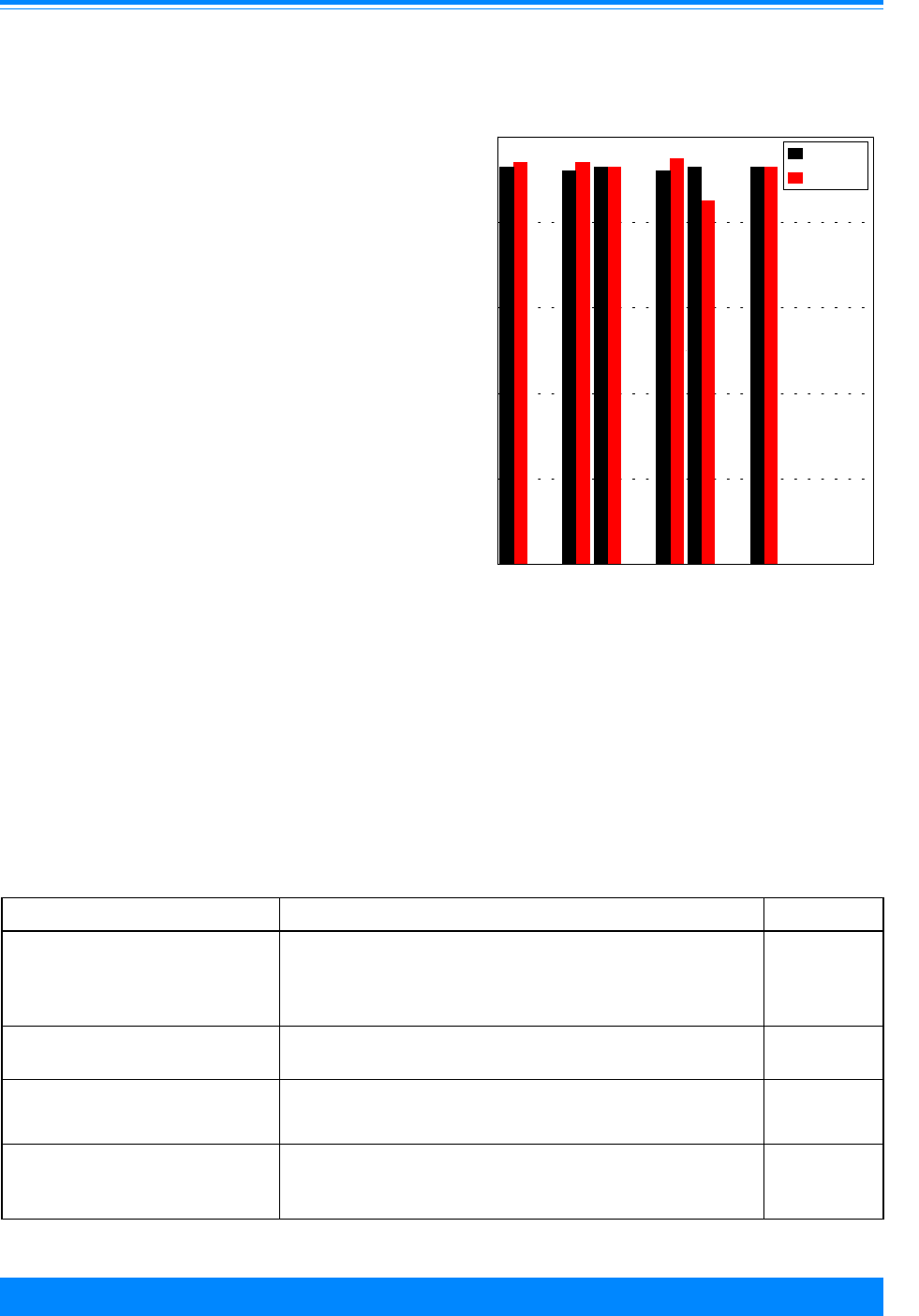
ICCC VRETINOBLASTOMA
77
National Cancer Institute SEER Pediatric Monograph
RISK FACTORS
While the genetics of retinoblastoma
are well understood, there is much less
known about the role of non-genetic factors
in retinoblastoma (Table V.3). Each child of
a parent with familial bilateral retinoblas-
toma has a 50% risk of inheriting the
retinoblastoma gene. Patients with spo-
radic heritable retinoblastoma carry the
gene for retinoblastoma and can also pass
the gene on to their children even though
they did not inherit the gene from their
parents. Children who inherit the retino-
blastoma gene have a 90% risk of develop-
ing retinoblastoma. Sporadic
(nonheritable) retinoblastoma results from
post-conception events and has been associ-
ated in a single study with parental occupa-
tion (Table V.3) [9].
SUMMARY
Retinoblastomas occur among very
young children, usually before the age of
five years. The incidence is about equal
among males and females and among black
children and white children. Rates have
changed little over the 21-year period,
Figure V.5: Retinoblastoma 5-year relative survival
rates, by sex, race, age and time period
SEER (9 areas), 1975-84 and 1985-94
# - <25 cases - rate not shown
93
92
93
92
93 93
94 94
93
95
85
93
Total Male Female White Black <5 5-9 10-1415-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex Race
Age
###
Known risk factors
Factors for which evidence is
inconsistent or limited
Table V.3: Current knowledge of causes of retinoblastoma
Exposure or Characteristic Comments References
Parent with history of bilateral
retinoblastoma
Each child has a 50% risk of inheriting the retinoblastoma
gene. If the gene is inherited, the risk of retinoblastoma is
over 90%. A small proportion of unilateral patients also
carry the gene and can pass it on to their children.
4,10
13q deletion syndrome Recognition of this syndrome led to the identification of the
retinoblastoma gene.
10
Paternal occupation There is a single report of association with employment in
the military, metal manufacturing, and as welder,
machinist, or related occupation.
9

ICCC V RETINOBLASTOMA
78
National Cancer Institute
SEER Pediatric Monograph
1975-95. Familial and sporadic heritable
retinoblastomas are caused by genetic
mutations and both usually result in the
bilateral form of the disease. Little is
known about the causes of sporadic non-
heritable retinoblastomas. Survival rates
are greater than 90% for children with
retinoblastoma.
Reference List
1. Donaldson S, Egbert P, Newsham I, et al:
Retinoblastoma. In Principles and Practice of
Pediatric Oncology (Pizzo P, Poplack D, eds).
Philadelphia: Lippincoltt-Raven, 1997, pp 699-
715.
2. Dryja TP, Morrow JF, Rapaport JM: Quantifi-
cation of the paternal allele bias for new
germline mutations in the retionoblastoma
gene. Hum genet 100:446-9, 1997.
3. Dryja TP, Mukai S, Pertersen R, et al: Parental
origin of mutations of the retinoblastoma gene.
Nature 339:556-8, 1989.
4. Knudson Ag, Jr.: Mutation and cancer: statisti-
cal study of retinoblastoma. Proc Natl Acad Sci
U S A 68:820-3, 1971.
5. Knudson AG: Antioncogenes and human
cancer. Proc Natl Acad Sci U S A 90:10914-21,
1993.
6. Dryja TP, Rapaport JM, Joyce JM, et al:
Molecular detection of deletions involving band
q14 of chromosome 13 in retinoblastomas. Proc
Natl Acad Sci U S A 83:7391-4, 1986.
7. Friend, SH, Bernards R, Rogelj S, et al: A
human DNA segment with properties of the
gene that predisposes to retinoblastoma and
osteosarcoma. Nature 323:643-6, 1986.
8. Weinberg RA: The retinoblastoma protein and
cell cycle control. Cell 81:323-30, 1995.
9. Bunin GR, Petrakova A, Meadows AT, et al:
Occupations of parents of children with
retinoblastoma: a report from the Children’s
Cancer Study Group. Cancer Res 50:7129-33,
1990.
10. Li F: Familial aggregation. In Cancer Epidemi-
ology and Prevention (Schottenfeld D,
Fraumeni J, eds). New York: Oxford University
Press, 1996, pp 546-558.

ICCC VI
79
RENAL TUMORS
79
National Cancer Institute
79
National Cancer Institute
SEER Pediatric Monograph
79
National Cancer Institute
SEER Pediatric Monograph
Leslie Bernstein, Martha Linet, Malcolm A. Smith, Andrew F. Olshan
HIGHLIGHTS
Incidence
♦ Malignancies of the kidney (renal cancers) represented 6.3% of cancer diagnoses
among children younger than 15 years of age (incidence 7.9 per million) (Table
VI.2) and 4.4% of cancer diagnoses for children and adolescents younger than 20
years of age (incidence of 6.2 per million).
♦ In the US approximately 550 children and adolescents younger than 20 years of
age are diagnosed with renal tumors each year, of which approximately 500 are
Wilms’ tumor.
♦ Wilms’ tumor was by far the most common form of renal cancer in children
younger than 15 years of age, representing approximately 95% of diagnoses
(Tables VI.1 and VI.2). Much less common were rhabdoid tumors of the kidney
(1% of renal cancers) and clear cell sarcoma of the kidney (1.6% of renal cancers).
Renal carcinomas, the most common form of renal cancer in adults, represented
only 2.6% of renal cancers in children younger than 15 years of age.
♦ Wilms’ tumor occurred most commonly among children younger than 5 years of
age (Figure VI.1), with very low incidence for 10-14 and 15-19 year olds. The
highest incidence for Wilms’ tumor was in the first 2 years of life, followed by
steadily decreasing rates with increasing age (Figure VI.2).
♦ Rhabdoid tumor of the kidney was diagnosed primarily in infants, while clear cell
sarcoma of the kidney was diagnosed primarily during the first 4 years of life.
Renal carcinomas, by contrast, occurred with highest incidence among 15-19 year
olds (Figure VI.1).
♦ Females had slightly higher incidence than males for Wilms’ tumor during the
period 1975-95 (Table VI.3). For the recent period of 1990-95, however, incidence
rates were similar by sex (Figure VI.3).
♦ Black children had somewhat higher incidence for Wilms’ tumor than white chil-
dren for the period 1975-95. For the time periods 1986-89 and 1990-95, however,
incidence rates by race were similar (Figure VI.4).
♦ Incidence of Wilms’ tumor showed neither substantial increases nor decreases
during the 21-year period from 1975 to 1995 (FigureVI.5).
Survival
♦ The overall relative 5-year survival rate for children with Wilms’ tumor was ap-
proximately 92% for cases diagnosed from 1985-94 (Figure VI.6), an improvement
from the 81% survival rate for cases diagnosed from 1975-84. Relative survival
rates were slightly higher for females than males and slightly higher for black
children than for white children (Figure VI.6).
Risk factors
♦ Certain congenital anomalies and genetic conditions increase susceptibility for
Wilms’ tumor (Table VI.4). Suggestive, although not conclusive, data indicate that
certain paternal occupations may be associated with increased Wilms’ tumor risk.

ICCC VI RENAL TUMORS
80
National Cancer Institute SEER Pediatric Monograph
INTRODUCTION
Renal tumors occurring in children
comprise a spectrum of morphologic sub-
types, including some with benign histopa-
thology. Wilms’ tumor (also called
nephroblastoma or renal embryoma) is by
far the most common form of renal cancer
in children. Other rarer forms of childhood
renal cancers are: clear cell sarcoma of the
kidney, rhabdoid tumor of the kidney,
congenital mesoblastic nephroma, mul-
tilocular cystic renal tumor, renal cell
carcinoma, and angiomyolipoma [1,2].
During 1975-95 in regions covered by
SEER cancer registries, malignant forms of
renal tumors represented 6.3% of total
cancer diagnoses among children younger
than 15 years of age and 4.4% for the
younger than 20 years old population. The
contribution of renal cancers to the overall
childhood cancer burden was notably age-
dependent, with renal cancers representing
9.7% of total incident malignancies diag-
nosed among children younger than 5 years
of age, 5.4% in children 5-9 years of age,
1.1% in children 10-14 years of age, and
only 0.6% in adolescents 15-19 years of age.
Wilms’ tumor is believed to arise from
primitive metanephric blastema (i.e., the
tissue from which the normal kidney
arises), though this tumor type often con-
tains tissues not occurring in the develop-
ing kidney, including skeletal muscle,
cartilage, and squamous epithelium [3].
Wilms’ tumor usually arises in only one of
the affected child’s kidneys, although
approximately 12% of affected children may
be diagnosed with Wilms’ tumor that is
multicentric in origin [4]. Approximately
7% of children with Wilms’ tumor have
involvement of both kidneys. Patients with
bilateral forms are generally diagnosed at
younger ages and are more likely to have
associated developmental abnormalities
than patients with unilateral forms [4]. In
the US approximately 550 children and
adolescents younger than 20 years of age
are diagnosed with renal tumors each year,
of which approximately 500 are Wilm’s
tumor.
Classification System
The International Classification for
Childhood Cancers (ICCC) Group VI of
Renal Cancers divides malignant neo-
plasms into three subgroups [5]:
a. Wilms’ tumor, rhabdoid tumor of
the kidney, and clear-cell sar-
coma of the kidney
b. Renal carcinoma
c. Unspecified malignant renal
tumors.
Table VI.1: Number of cases and percent distribution of renal cancers by histologic
subtype and age group, all races, both sexes, SEER, 1975-95
Age (in years) at diagnosis <5 5-9 10-14 15-19 <15 <20
Wilms' tumor 880 260 39 21 1,179 1,200
(96.2%)
1
(95.9%) (66.1%) (35.0%) (94.7%) (92.0%)
Rhabdoid tumor of the kidney 12 * * * 12 12
(1.3%) (1.0%) (0.9%)
Clear cell sarcoma of the kidney 16 * * * 19 19
(1.8%) (1.6%) (1.6%)
Renal carcinoma
6 7 19 38 32 70
(0.7%) (2.6%) (32.2%) (63.3%) (2.6%) (5.4%)
Unspecified renal cancer
******
Total renal cancers 915 271 59 60 1,245 1,305
1
Number in parenthesis represents the percentage of all renal cancers for the age group that are
represented by the histologic category.
*Less than 5 cases.

81
National Cancer Institute SEER Pediatric Monograph
ICCC VIRENAL TUMORS
81
National Cancer Institute
SEER Pediatric Monograph
The numbers of cases of these histologic
diagnoses among children residing in the
SEER areas for the period 1975-95 are
shown in Table VI.1. Malignant forms of
renal tumors were diagnosed in 1,245
children younger than 15 years of age and
in 1,305 children younger than 20 years of
age. Wilms’ tumor was by far the most
common form of renal cancer, accounting
for 94.7% of the 1,245 renal cancers in
children younger than 15 years of age and
92.0% of the 1,305 renal cancers among the
younger than 20 year olds. Occurring much
less commonly among the total 1,245 cases
of renal cancer in children younger than 15
years of age were rhabdoid tumor of the
kidney (12 cases representing 1.0% of renal
cancers) and clear cell sarcoma of the
kidney (19 cases representing 1.6% of renal
cancers). For renal cell carcinoma, there
were 32 cases among children younger than
15 years of age (2.6% of renal cancers) and
70 cases among children younger than 20
years of age (5.4% of renal cancers).
Wilms’ tumor, rhabdoid tumor of the
kidney, and clear cell sarcoma of the kidney
are classified together in the ICCC category
VIa, while the renal carcinomas are
grouped together in the ICCC category VIb.
In presenting incidence data, the ICCC
category VIa (for which Wilms’ tumor
represents greater than 95% of cases for all
age groups) is designated Wilms’ tumor
(ICCC VIa), since the incidence patterns
and trends for this subcategory are largely
determined by cases of Wilms’ tumor.
When Wilms’ tumor is discussed as a single
diagnosis, the term Wilms’ tumor without
any parenthetic modifier is used. Addition-
ally, since incidence rates for renal cancers
were low and based on very small numbers
in children ages 15-19 (a total of 60 inci-
dent cases diagnosed in the SEER areas
during 1975-95), presentation of incidence
data are generally restricted to renal
cancers diagnosed among children younger
than 15 years of age.
INCIDENCE
The average annual age-adjusted
incidence rates of renal cancer for the years
1975-95 was 7.9 per million for children
younger than 15 years of age (Table VI.2)
and 6.2 per million for children younger
than 20 years of age in the SEER areas. As
discussed in greater detail in subsequent
paragraphs, incidence rates for renal can-
cers in toto and for Wilms’ tumor (ICCC
VIa) for the period 1975-95 were slightly
higher for females than males and for black
children compared to white children. How-
ever, for the most recent time period (1990-
95), rates were similar for both sexes and
for black children and white children.
Age-specific incidence
Age-specific incidence rates of renal
cancers in 5-year age groups were highest
among children younger than 5 years of age
Table VI.2: Age-adjusted* incidence rates for renal cancer
by race and sex, age <15, SEER, 1975-95
Both sexes Males Females
Renal cancers (VI)
All races 7.9 7.4 8.4
Whites 8.0 7.4 8.6
Blacks 9.5 9.5 9.6
Wilms’ tumor (VIa)
All races 7.6 7.1 8.1
Whites 7.9 7.3 8.4
Blacks 8.7 8.4 9.0
*Adjusted to the 1970 US standard population

ICCC VI RENAL TUMORS
82
National Cancer Institute SEER Pediatric Monograph
(Table VI.3). Incidence declined markedly
with increasing age. The age-incidence
pattern for renal cancer in children was
driven by that for Wilms’ tumor (ICCC
VIa), as illustrated in Figure VI.1. Renal
cell carcinomas (ICCC VIb) occurred very
infrequently among each 5-year age group
younger than 15 years of age, but for 15-19
year olds the incidence rate was higher
(though still only 0.7 per million) (Figure
VI.1). Among the 15-19 year old popula-
tion, renal carcinomas represented the
majority (63%) of cases of renal cancer.
Average annual incidence rates for
Wilms’ tumor by single year of age are
presented in Figure VI.2 for the time peri-
ods 1976-84 and 1986-94.
1
The age-specific
incidence rates were highest in the first two
years of life at 21 per million, with inci-
dence rates subsequently declining to levels
less than 2 per million for children older
than 9 years of age. Age-specific rates for
the other renal cancers were much lower
than those for Wilms’ tumor. Rhabdoid
tumors of the kidney was present almost
exclusively in the first 2 years of life, with a
peak in infancy of 1.0 per million. Clear
cell sarcoma of the kidney also occurred
much less frequently than Wilms’ tumor,
with age-specific incidence in the first 4
years of life ranging between 0.4 and 0.6
per million, and with very few cases occur-
ring among children older than 3 years of
age. Renal cell carcinoma was also uncom-
mon among children of any age, with most
cases occurring in adolescents 15-19 years
of age, for which age-specific incidence rates
varied between 0.5 and 0.9 per million.
Sex-specific incidence
For the 21 year period from 1975 to
1995, renal cancer incidence rates among
children younger than 15 years of age were
minimally higher for females compared to
males (13 percent higher among females)
(Table VI.2). For Wilms’ tumor (ICCC VIa),
there was also a slight female
predominance when the overall period
1975-95 was considered. However, inci-
dence rates for Wilms’ tumor (ICCC VIa)
were the same for males and females for
the most recent period evaluated (1990-95)
(Figure VI.3). As illustrated in Figure VI.2,
females had slightly higher rates of Wilms’
tumor than males in infancy (22.6 per
Figure VI.1: Renal cancer age-specific
incidence rates by ICCC subcategory,
all races, both sexes, SEER, 1975-95
18.3
5.6
0.8
0.4
0.1
0.1
0.4
0.7
<5 5-9 10-14 15-19
Age (in years) at diagnosis
0
5
10
15
20
Average annual rate per million
Wilms (VIa)
Renal carcinoma (VIb)
Table VI.3: Age-specific incidence rates per
million for renal cancer by age
and race, SEER, 1975-95
All races Whites Blacks
Males
<5 years 17.8 18.2 21.5
5-9 years 4.9 4.9 6.7
10-14 years 1.2 1.1 2.2
Females
<5 years 19.1 19.6 21.4
5-9 years 6.6 6.8 7.8
10-14 years 1.2 1.3 1.6
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.

83
National Cancer Institute SEER Pediatric Monograph
ICCC VIRENAL TUMORS
83
National Cancer Institute
SEER Pediatric Monograph
Figure VI.3: Wilms' tumor (VIa) age-adjusted* incidence rates
by sex and year of diagnosis, age <15 all races, both sexes, SEER 1975-95
6.4
7.5
7.3 7.3
8.5
8.7
8.4
7.3
1975-79 1980-84 1985-89 1990-95
Year of Diagnosis
0
2
4
6
8
10
Average annual rate per million
Males
Females
*Adjusted to the 1970 US standard population
Figure VI.2: Wilms' tumor (VIa) age-specific incidence rates
by sex, all races, SEER, 1976-84 and 1986-94
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
0123456789101112131415
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
Males
Females
Total
"
)
#

ICCC VI RENAL TUMORS
84
National Cancer Institute SEER Pediatric Monograph
million versus 19.7 per million, respec-
tively). Among children 3-8 years old, the
age-specific incidence rates for females
were generally equal to or greater than
rates for males. For females, but not for
males, the steady decline in Wilms’ tumor
incidence rates with increasing age after
infancy was apparent except in the fourth
year of life during which rates increased in
females to levels approaching those seen in
infancy, then subsequently declined lin-
early.
Black-white differences in incidence
Renal cancer and Wilms’ tumor (ICCC
VIa) incidence rates for the overall period
1975-95 were somewhat higher for black
children than for white children (Table
VI.2). For the two most recent five year
periods (1985-89 and 1990-95), however,
incidence rates for Wilms’ tumor were
similar for black children and white chil-
dren (Figure VI.4).
TRENDS
The age-adjusted incidence rates for
childhood renal cancers did not change
significantly during the period 1975-95.
Incidence rates for Wilms’ tumor (ICCC
VIa) varied from year to year (Figure VI.5),
but there was no trend for increase or
decrease during the 21-year period (esti-
mated annual percentage change = -0.03%).
The incidence of renal carcinoma was very
low throughout the period (Figure VI.5).
For the years 1975-79, Wilms’ tumor (ICCC
VIa) incidence rates for females (8.5 per
million) were higher than for males (6.4 per
million) (Figure VI.3). However, rates for
males rose between 1975-79 and 1980-84 to
7.5 per million, and thereafter remained
fairly stable. Rates for females declined,
particularly between 1985-89 and 1990-95,
so that females and males had the same
incidence rate (7.3 per million) during
1990-95 (Figure VI.3). Incidence rates for
Wilms’ tumor (ICCC VIa) for white children
did not vary much between each 5-6 year
time period from 1975 to 1995. Black
Figure VI.4: Wilms' tumor (VIa) age-adjusted* incidence
rates by race and year of diagnosis, age <15
both sexes, SEER, 1975-95
7.6
7.9
8.2
7.8
8.5
11.7
8.2
7.1
1975-79 1980-84 1985-89 1990-95
Year of diagnosis
0
2
4
6
8
10
12
14
Average annual rate per million
White
Black
*Adjusted to the 1970 US standard population
Figure VI.5: Trends in renal cancer age-adjusted*
incidence rates by type, age <15
all races, both sexes, SEER, 1975-95
%
%
%%
%
%
%
%
%
%
%
%
%
%%
%
%
%
%
%
%
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
'
1975 1980 1985 1990 1995
Year of diagnosis
0
2
4
6
8
10
12
Average annual rate per million
Wilms' Tumor (VIa)
Renal Carcinoma (VIb)
'
%
*Adjusted to the 1970 US standard population

85
National Cancer Institute SEER Pediatric Monograph
ICCC VIRENAL TUMORS
85
National Cancer Institute
SEER Pediatric Monograph
children had higher incidence rates for
Wilms’ tumor (ICCC VIa) in 1975-79 and
1980-84 (8.5 and 11.7 per million, respec-
tively), but the rates dropped for the years
1985-89 and 1990-95 (8.2 and 7.1 per
million, respectively) to levels very similar
to those for white children (Figure VI.4).
SURVIVAL
For children of all ages, both sexes, and
all racial/ethnic groups residing in the
SEER areas, the relative 5-year survival
rate for children diagnosed with Wilms’
tumor younger than 15 years of age during
1985-94 was 92%, compared with a rate of
81% among children diagnosed with this
malignancy during 1975-84. Among cases
diagnosed during 1985-94, 5-year survival
was slightly better for females (94%) than
males (91%). For 1985-94, black children
had somewhat better outcome than white
children (95% versus 92% 5-year survival)
(Figure VI.6). Children with rhabdoid
tumor are known to have a much poorer
outcome than children with Wilms’ tumor
[44], and among the small number of
children with rhabdoid tumor of the kidney
followed for survival in SEER areas (n = 8),
all either died (6) or were lost to follow-up
(2). Children with clear cell sarcoma of the
kidney are known to have a somewhat
poorer prognosis than children with Wilms’
tumor, with 6-year relapse-free survival
rates of slightly above 60% based on data
from the US National Wilms’ Tumor Study
Group [45,46]. There were only 13 children
with clear cell sarcoma of the kidney
evaluable for survival from the SEER areas
for the time period 1975-94, and their
relative 5-year survival rate was 84%. For
children and adolescents with renal carci-
nomas, 5-year relative survival rates in-
creased from 48% for cases diagnosed in
1975-84 to 83% for cases diagnosed in
1985-94 (although these estimates are
Figure VI.6: Wilms' tumor 5-year relative survival rates
by race and sex, age <15, SEER (9 Areas), 1975-84 and 1985-94
81
80
81
78
83
92 92
95
91
94
All White Black Males Females
0
20
40
60
80
100
Percent surviving 5 years
1975-84 1985-94
(
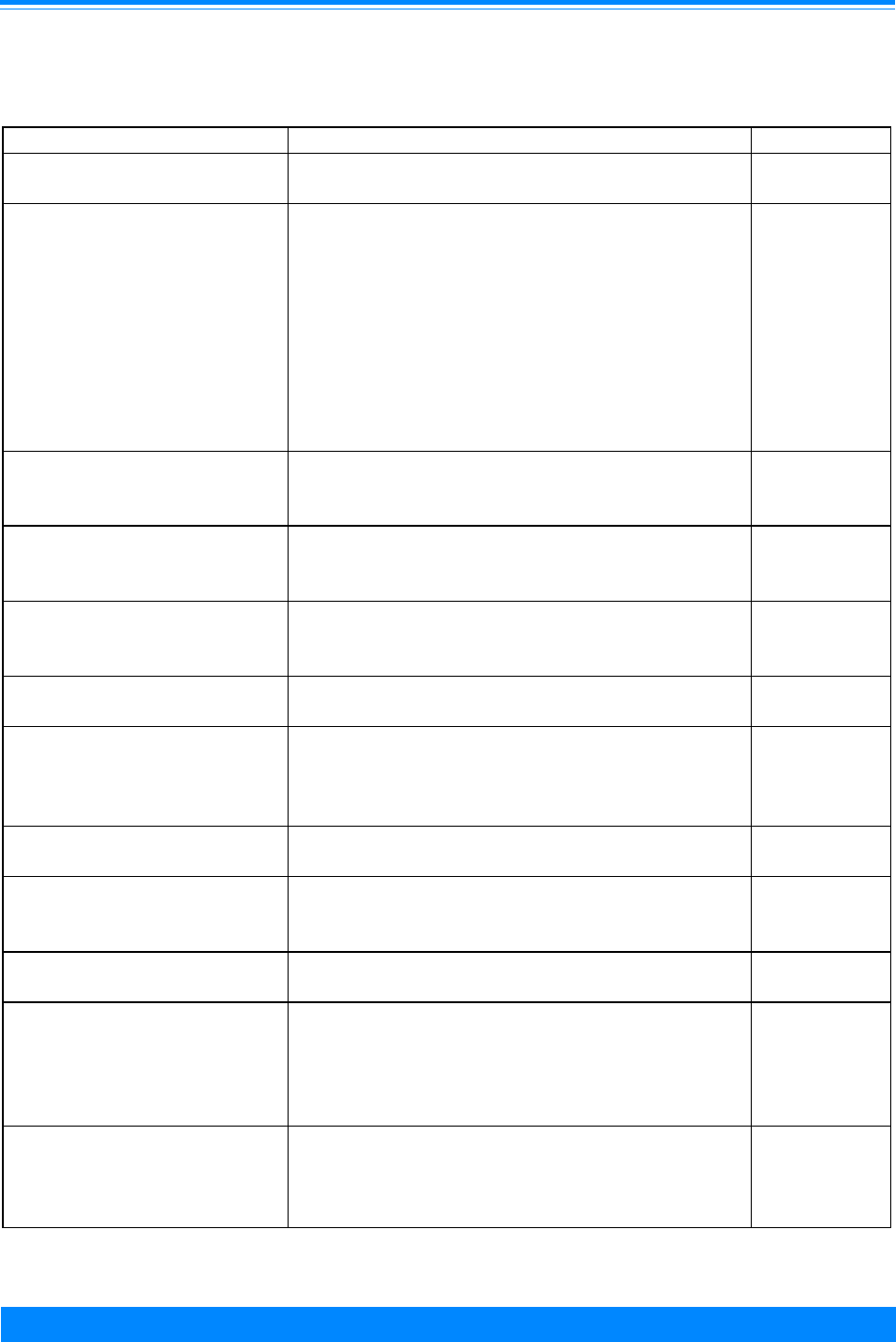
ICCC VI RENAL TUMORS
86
National Cancer Institute SEER Pediatric Monograph
Table VI.4: Current knowledge on causes of Wilms’ Tumor (WT)
Exposure or Characteristic Comments References
Race Incidence in Asians is about half that in blacks
and whites.
10,11
Aniridia, genitourinary
anomalies, WAGR
syndrome (Wilms’ tumor,
aniridia, genitourinary
abnormalities, mental
retardation), Beckwith-
Wiedemann syndrome,
Perlman syndrome, Denys-
Drash syndrome, Simpson-
Golabi-Behmel syndrome
Risk is increased in children with these
congenital anomalies and genetic conditions.
The study of children with WAGR led to the
identification of one of the WT genes.
12-22
Paternal occupation An increased risk for fathers employed as a
welder or mechanic has been reported in
several studies.
13,26,28
High birth weight Association with birth weight over 4,000 grams
has been reported in some studies.
13,29,30
Parental exposure to
pesticides
One study found an increased risk for parental
occupational exposure to pesticides. Another
study found an association with household
insect extermination.
13,27,31-33
Ionizing radiation (in utero) Prenatal diagnostic x-ray was associated with
increased risk in one study.
34
Maternal consumption of
coffee and tea during
pregnancy
Three studies reported association with coffee
and/or tea; another did not replicate this
finding.
31,35,36
Maternal hair dye use
during pregnancy
Use was associated with risk in one study, but
not in others.
31,36
Maternal medication use
during pregnancy
Studies reported associations with various
drugs including hormones, antibiotics,
dipyrone, metoclopramide, pethrane anesthesia
during delivery. Most of these results were
found in only a single study.
13,37,38
Maternal occupation One study found an association with job
groupings that included hairdressers,
electronic and clothing manufacturing workers,
laboratory workers, dental assistants.
26,39
Known risk factors
Factors for which
evidence is inconsistent
or limited
Factors for which
evidence is suggestive
but not conclusive

87
National Cancer Institute SEER Pediatric Monograph
ICCC VIRENAL TUMORS
87
National Cancer Institute
SEER Pediatric Monograph
based on only 23 and 29 cases, respectively,
from these two time periods).
RISK FACTORS
Despite the rarity of renal cancer in
children, a substantial body of epidemio-
logic, genetic, and molecular studies have
contributed important insights to under-
standing its pathogenesis [3,6,7]. Histori-
cally, Wilms’ tumor was thought to vary
little in incidence throughout the world and
was therefore proposed as an “index tumor”
of childhood cancer [8]. However, interna-
tional comparisons based on data through
the 1980s showed a greater than threefold
difference in age-adjusted incidence rates
among populations, with highest rates
observed in US and Nigerian blacks, fol-
lowed by somewhat lower rates in Sweden
and US whites, and lowest rates in Chinese
and other Asians [9,10]. Data for the years
1973-88 from the US showed similar ethnic
variation, with incidence in Asians about
half that in blacks, and rates for blacks
slightly higher than rates for whites [11]
(Table VI.4).
A small proportion of Wilms’ tumor
cases appear to be heritable including:
those patients with bilateral tumors, those
occurring in association with aniridia and
other congenital anomalies, and those few
cases arising in the small number of fami-
lies with one or more additional cases of
Wilms’ tumor in close family members
[12,13]. Approximately 1.5% of patients in
a large series had one or more family
members (usually siblings or cousins) with
Wilms’ tumor based on interview data [14].
Congenital disorders that have been linked
with Wilms’ tumor include: the Beckwith-
Wiedemann syndrome (an overgrowth
syndrome associated with macrosomia,
omphalocele, macroglossia, and viscerome-
galy and believed to be linked to an as yet
unidentified gene(s) at chromosome region
11p15) [14,15]; the Simpson-Golabi-Behmel
syndrome (an X-linked fetal overgrowth
disorder caused by mutations in the
glypican 3 gene) [15,16]; hemihypertrophy
as an isolated abnormality; the Perlman
and Sotos syndromes [17-19]; the Denys-
Drash syndrome (associated with muta-
tions of the Wilms’ tumor suppressor gene
WT1) [20-22]; and the WAGR syndrome
(Wilms’ tumor, aniridia, genitourinary
malformations, and mental retardation )
that results from deletion of a number of
contiguous genes on chromosome 11 includ-
ing the aniridia gene PAX6 and the WT1
gene [12]. In addition to the heritable
conditions cited above, inherited predisposi-
tion genes associated with some familial
Wilms’ tumor cases appear to exist at two
other loci (and possibly others not yet
identified) [23,24]. However, survivors of
Wilms’ tumor that is unilateral at diagnosis
are at low risk for having children with
Wilms’ tumor [25].
Most of the analytical and epidemio-
logic investigations of childhood renal
cancer have focused on Wilms’ tumor, and
very little is known about risk factors for
childhood renal carcinoma or the other
rarer childhood renal cancer subtypes.
Several epidemiological studies have inves-
tigated occupational, environmental, and
lifestyle characteristics as potential risk
factors for Wilms’ tumor, but findings to
date have been inconsistent [13,26,27]. A
few studies have suggested that children of
fathers employed as welders or mechanics
have increased risk of Wilms’ tumor [13,28],
but occupational exposure assessment was
insufficient to draw firm conclusions [26].
Limited evidence implicates high birth
weight in the etiology [29,30]. Parental
and postnatal exposures to pesticides have
also been linked with increased risk [27,31-
33], but these associations were derived
from interview data only and have not been
confirmed with studies utilizing measure-
ments. A large study in the United King-
dom has reported an association of Wilms’
tumor with exposure of the mother to
ionizing radiation from diagnostic x-rays

ICCC VI RENAL TUMORS
88
National Cancer Institute SEER Pediatric Monograph
during pregnancy [34.] Some [31,32,35],
but not all [36] investigations have found
associations between maternal consump-
tion of coffee and tea during pregnancy and
risk of Wilms’ tumor in offspring. Inconsis-
tent reports have also implicated maternal
hair dye use and various types of medica-
tions taken or anesthetics to which mothers
have been exposed during pregnancy
[13,37,38]. The role of maternal occupa-
tional exposures has received limited
evaluation [26,39].
Most of the reported associations
described in the preceding paragraph have
not been consistently replicated in multiple,
high quality studies in different popula-
tions. Future epidemiologic studies may
benefit from more detailed exposure assess-
ment, validated by environmental and
biologic measurements. In addition, the
role of genetic susceptibility and assess-
ment of gene-environment interaction
should be considered by evaluation of
appropriate molecular markers to better
define etiologic pathways for Wilms’ tumor.
Recurring molecular abnormalities
have been identified in the tumor cells of
two of the uncommon renal cancers that
occur in young children, rhabdoid tumor of
the kidney and congenital mesoblastic
nephroma. Rhabdoid tumor, which can
develop in the central nervous system and
extrarenal sites as well as in the kidney, is
associated with tumor cell mutations in the
INI1 gene located on chromosome 22
[40,41]. Evaluation of some children with
rhabdoid tumors has revealed germline
mutations of the INI1 gene [41]. Congeni-
tal mesoblastic nephroma is an infantile
spindle cell tumor of the kidney with low
malignant potential that is virtually identi-
cal morphologically to congenital fibrosar-
coma [42]. The tumor cells of both of these
tumors of infancy have been found to
possess fusions of the ETV6 gene (also
known as TEL) on chromosome 12 to the
NTRK3 gene on chromosome 15 [42,43].
SUMMARY
The descriptive epidemiologic features
of Wilms’ tumor have been known for a
number of years. Associated congenital
anomalies and genetic factors have also
been subject of much interest. More recent
studies have further characterized the
specific genetic loci and molecular alter-
ations involved in the development of
Wilms’ tumor. Several epidemiologic stud-
ies have investigated occupational, environ-
mental, and lifestyle factors as risk factors
for Wilms’ tumor. A number of parental
and childhood exposures have been found
to be associated with an increased risk of
Wilms’ tumor. Most of these associations
have not been replicated in multiple high
quality studies. However, some warrant
further evaluation including paternal
occupational exposures, pesticide exposure,
and certain maternal exposures during
pregnancy. Future epidemiologic studies
may benefit from the inclusion of molecular
markers that may better define etiologic
pathways for Wilms’ tumor.
Reference List
1. Charles AK, Vujanic GM, Berry PJ: Renal
tumours of childhood. Histopathology 32:293-
309, 1998.
2. Geller E, Smergel EM, Lowry PA: Renal
neoplasms of childhood. Radiol Clin North Am
35:1391-413, 1997.
3. Green D, Coppes M, Breslow N, et al: Wilms’
tumor. In Principles and Practice of Pediatric
Oncology (Pizzo P, Poplack D, eds). Philadel-
phia: Lippincott-Raven Publishers, 1997, pp
733-59.
4. Breslow N, Beckwith JB, Ciol M, et al: Age
distribution of Wilms’ tumor: Report from the
National Wilms’ Tumor Study. Cancer Res
48:1653-7, 1988.
5. Kramarova E, Stiller CA: The international
classification of childhood cancer. Int J Cancer
68:759-65, 1996.
6. Huff V, Amos CI, Douglass EC, et al: Evidence
for genetic heterogeneity in familial Wilms’
tumor. Cancer Res 57:1859-62, 1997.
7. Rahman N, Arbour L, Tonin P, et al: Evidence
for a familial Wilms’ tumour gene (FWT1) on
chromosome 17q12-q21. Nat Genet 13:461-3,
1996.

89
National Cancer Institute SEER Pediatric Monograph
ICCC VIRENAL TUMORS
89
National Cancer Institute
SEER Pediatric Monograph
8. Innis MD: Wilms’ tumour. Med J Aust 1:282,
1972.
9. Parkin D, Stiller C, Draper G, et al: Interna-
tional Incidence of Childhood Cancer. Lyon:
World Health Organization, 1988.
10. Stiller CA, Parkin DM: International varia-
tions in the incidence of childhood renal
tumours. Br J Cancer 62:1026-30, 1990.
11. Breslow N, Olshan A, Beckwith JB, et al:
Ethnic variation in the incidence, diagnosis,
prognosis, and follow-up of children with
Wilms’ tumor. J Natl Cancer Inst 86:49-51,
1994.
12. Coppes MJ, Haber DA, Grundy PE: Genetic
events in the development of Wilms’ tumor. N
Engl J Med 331:586-90, 1994.
13. Sharpe CR, Franco EL: Etiology of Wilms’
tumor. Epidemiol Rev 17:415-32, 1995.
14. Beckwith J: Macroglossia, omphalocele,
adrenal cytomegaly, gigantism and hyperplas-
tic visceromegaly. Birth Defects 5:188-, 1969.
15. Li M, Squire JA, Weksberg R: Overgrowth
syndromes and genomic imprinting: from
mouse to man. Clin Genet 53:165-70, 1998.
16. Neri G, Gurrieri F, Zanni G, et al: Clinical and
molecular aspects of the Simpson-Golabi-
Behmel syndrome [In Process Citation]. Am J
Med Genet 79:279-83, 1998.
17. Miller R, Fraumeni JJ, Manning M: Associa-
tion of Wilms’ tumor with aniridia, hemihyper-
trophy and other congenital malformations. N
Engl J Med 270:922-927, 1964.
18. Perlman M, Levin M, Wittels B: Syndrome of
fetal gigantism, renal hamartomas, and
nephroblastomatosis with Wilms’ tumor.
Cancer 35:1212-7, 1975.
19. Hersh JH, Cole TR, Bloom AS, et al: Risk of
malignancy in Sotos syndrome [see comments].
J Pediatr 120:572-4, 1992.
20. Denys P, Malvaux P, Van Den Berghe H, et al:
[Association of an anatomo-pathological
syndrome of male pseudohermaphroditism,
Wilms’ tumor, parenchymatous nephropathy
and XX/XY mosaicism]. Arch Fr Pediatr
24:729-39, 1967.
21. Drash A, Sherman F, Hartmann WH, et al: A
syndrome of pseudohermaphroditism, Wilms’
tumor, hypertension, and degenerative renal
disease. J Pediatr 76:585-93, 1970.
22. Coppes MJ, Huff V, Pelletier J: Denys-Drash
syndrome: relating a clinical disorder to
genetic alterations in the tumor suppressor
gene WT1. J Pediatr 123:673-8, 1993.
23. Rahman N, Abidi F, Ford D, et al: Confirmation
of FWT1 as a Wilms’ tumour susceptibility
gene and phenotypic characteristics of Wilms’
tumour attributable to FWT1. Hum Genet
103:547-56, 1998.
24. McDonald JM, Douglass EC, Fisher R, et al:
Linkage of familial Wilms’ tumor predisposi-
tion to chromosome 19 and a two-locus model
for the etiology of familial tumors. Cancer
Research 58:1387-90, 1998.
25. Li FP, Williams WR, Gimbrere K, et al: Heri-
table fraction of unilateral Wilms’ tumor.
Pediatrics 81:147-9, 1988.
26. Colt JS, Blair A: Parental occupational expo-
sures and risk of childhood cancer [In Process
Citation]. Environ Health Perspect 106 Suppl
3:909-25, 1998.
27. Zahm SH, Ward MH: Pesticides and childhood
cancer. Environ Health Perspect 106 Suppl
3:893-908, 1998.
28. Breslow N, Olshan A, Beckwith JB, et al:
Epidemiology of Wilms’ tumor. Med Pediatr
Oncol 21:172-81, 1993.
29. Leisenring WM, Breslow NE, Evans IE, et al:
Increased birth weights of National Wilms’
Tumor Study patients suggest a growth factor
excess. Cancer Res 54:4680-3, 1994.
30. Yeazel MW, Ross JA, Buckley JD, et al: High
birth weight and risk of specific childhood
cancers: a report from the Children’s Cancer
Group. J Pediatr 131:671-7, 1997.
31. Olshan AF, Breslow NE, Falletta JM, et al:
Risk factors for Wilms’ tumor. Report from the
National Wilms’ Tumor Study. Cancer 72:938-
44, 1993.
32. Sharpe CR, Franco EL, de Camargo B, et al:
Parental exposures to pesticides and risk of
Wilms’ tumor in Brazil. Am J Epidemiol
141:210-7, 1995.
33. Fear NT, Roman E, Reeves G, et al: Childhood
cancer and paternal employment in agricul-
ture: the role of pesticides. Br J Cancer 77:825-
9, 1998.
34. Bithell JF, Stewart AM: Pre-natal irradiation
and childhood malignancy: a review of British
data from the Oxford Survey. Br J Cancer
31:271-87, 1975.
35. Le Masters G, Bove K: Genetic/environmental
significance of multifocal nodular renal
blastema. Am J Pediatr Hematol Oncol 2:81-
87, 1980.
36. Bunin GR, Kramer S, Marrero O, et al: Gesta-
tional risk factors for Wilms’ tumor: results of
a case-control study. Cancer Res 47:2972-7,
1987.
37. Lindblad P, Zack M, Adami HO, et al: Maternal
and perinatal risk factors for Wilms’ tumor: a
nationwide nested case-control study in
Sweden. Int J Cancer 51:38-41, 1992.
38. Sharpe CR, Franco EL: Use of dipyrone during
pregnancy and risk of Wilms’ tumor. Brazilian
Wilms’ Tumor Study Group. Epidemiology
7:533-5, 1996.

ICCC VI RENAL TUMORS
90
National Cancer Institute SEER Pediatric Monograph
39. Bunin GR, Nass CC, Kramer S, et al: Parental
occupation and Wilms’ tumor: results of a case-
control study. Cancer Res 49:725-9, 1989.
40. Versteege I, Sevenet N, Lange J, et al: Truncat-
ing mutations of hSNF5/INI1 in aggressive
paediatric cancer. Nature 394:203-6, 1998.
41. Biegel JA, Zhou JY, Rorke LB, et al: Germ-line
and acquired mutations of INI1 in atypical
teratoid and rhabdoid tumors. Cancer Res
59:74-9, 1999.
42. Knezevich SR, Garnett MJ, Pysher TJ, et al:
ETV6-NTRK3 gene fusions and trisomy 11
establish a histogenetic link between meso-
blastic nephroma and congenital fibrosarcoma.
Cancer Res 58:5046-8, 1998.
43. Knezevich SR, McFadden DE, Tao W, et al: A
novel ETV6-NTRK3 gene fusion in congenital
fibrosarcoma. Nat Genet 18:184-7, 1998.
44. Weeks DA, Beckwith JB, Mierau GW, et al:
Rhabdoid tumor of kidney. A report of 111
cases from the National Wilms’ Tumor Study
Pathology Center. Am J Surg Pathol 13:439-58,
1989.
45. Green DM, Breslow NE, Beckwith JB, et al:
Treatment of children with clear-cell sarcoma
of the kidney: a report from the National
Wilms’ Tumor Study Group. Journal of Clinical
Oncology 12:2132-7, 1994.
46. Green DM, Breslow NE, Beckwith JB, et al:
Comparison between single-dose and divided-
dose administration of dactinomycin and
doxorubicin for patients with Wilms’ tumor: a
report from the National Wilms’ Tumor Study
Group. J Clin Oncol 16:237-45, 1998.

ICCC VII
HEPATIC TUMORS
Marc Bulterys, Marc T. Goodman, Malcolm A. Smith, Jonathan D. Buckley
91
National Cancer Institute
SEER Pediatric Monograph
HIGHLIGHTS
Incidence
♦ Primary neoplasms of the liver are rare in children, comprising only 1.1% of malig-
nancies for children younger than 20 years of age. In the US, 100-150 children are
diagnosed with liver cancer each year.
♦ Primary liver cancer is subdivided into the following histologic subtypes:
hepatoblastoma comprises over two-thirds of the malignant tumors of the liver in
children and adolescents (79% <15 years of age; 66% <20 years of age) and
hepatocellular carcinoma accounts for most of the remaining cases. Hepatoblastoma
occurs primarily in children younger than 5 years of age while hepatocellular
carcinoma occurs primarily after 10 years of age (Figure VII.2).
♦ The rate of hepatoblastoma was highest among infants with rates rapidly declining
with increasing age (Figure VII.3). In contrast, the incidence of hepatocellular
carcinoma increased as age increased (Figure VII.2).
♦ For those younger than 20 years of age, there was little change in liver cancer
incidence during the 21-year period, with rates between 1.4 and 1.6 per million
throughout the time period (Table VII.1).
♦ The incidence of hepatoblastoma for children younger than 15 years of age increased
during the 1975-95 period while the incidence of hepatocellular carcinoma decreased
during the same period (Figure VII.4).
Survival
♦ Five-year survival rates for children with hepatoblastoma improved from 51% to
59% between 1976-84 and 1985-94 (Figure VII.5). Survival rates were substantially
lower for children and adolescents with hepatocellular carcinoma, with an improve-
ment in 5-year survival rates from 31% for the years 1976-84 to 42% for the years
1985-94 (Figure VII.5).
Risk factors
♦ The etiology of hepatoblastoma is as yet unknown but there are some tantalizing
clues (Table VII.2).
INTRODUCTION
Primary neoplasms of the liver are rare
in children, comprising only 1.1% of malig-
nancies in SEER areas for children younger
than 20 years of age. The ICCC category
for liver cancers (VII) is subdivided into the
following histologic subtypes:
hepatoblastoma (VIIa), hepatic carcinomas
(hepatocellular carcinoma) (VIIb), and
“unspecified” tumors of the liver (VIIc) [1].
In the US, 100-150 children younger than
20 years of age are diagnosed with hepatic
tumors each year. Hepatoblastoma com-
prises over two-thirds of the malignant
tumors of the liver in children (79%
younger than 15 years of age; 66% younger
than 20 years of age) and hepatocellular
carcinoma accounts for most of the remain-
ing cases. Most patients with
hepatoblastoma are younger than 4 years
of age at diagnosis, while hepatocellular
carcinoma occurs primarily after 10 years
of age.

ICCC VII
92
National Cancer Institute
SEER Pediatric Monograph
HEPATIC TUMORS
INCIDENCE
During the 21-year period from 1975
through 1995, there were 316 children
younger than 20 years of age in SEER
areas who were diagnosed with a primary
liver cancer, with 262 (83%) of these chil-
dren being younger than 15 years of age at
the time of diagnosis. Figure VII.1 shows
that the majority of these cancers were
hepatoblastomas, with the remainder being
almost exclusively hepatocellular carcino-
mas. For the entire 21-year period,
hepatoblastoma represented 79% of the
liver cancers for the younger than 15 year
age group, although for the most recent 6-
year period (1990-95) hepatoblastoma
accounted for an even higher proportion
(90%) of childhood liver cancers.
Age-specific incidence
The incidence rates for hepatoblastoma
and hepatocellular carcinomas were very
age-dependent. Among children younger
than 5 years of age, over 95% of liver
cancers were hepatoblastoma, whereas
hepatoblastoma was distinctly uncommon
for older age groups (Figure VII.2). Within
the younger than 5-year age group, the rate
of hepatoblastoma was highest among
infants with rates rapidly declining with
increasing age (Figure VII.3).
1
During the
most recent period (1986-94), the incidence
rate during infancy was approximately 11.2
per million. In contrast to the age-inci-
dence relationship for hepatoblastoma, the
incidence of hepatocellular carcinoma
increased with each successive 5-year age
group, with rates for 15-19 year olds (0.9
per million) being substantially higher than
for any of the younger age groups (Figure
VII.2).
Figure VII.1: Distribution of liver cancer
by histology and age, all races
both sexes, SEER 1975-95
79
21
0.4
66
33
1
Hepatoblastoma
Hepatocellular
Other
0 20406080100
Percent of total liver cancer
<15 years
<20 years
carcinoma
Figure VII.2: Liver cancer age-specific
incidence rates by histology and age
all races, both sexes, SEER, 1986-95
4.6
0.2
0.1
0.04
0.2
0.2
0.3
0.9
<5 5-9 10-14 15-19
Age in years
0
1
2
3
4
5
6
Average annual rate per million
Hepatoblastoma
Hepatocellular carcinoma
4.8
0.4
0.4
1.0
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.
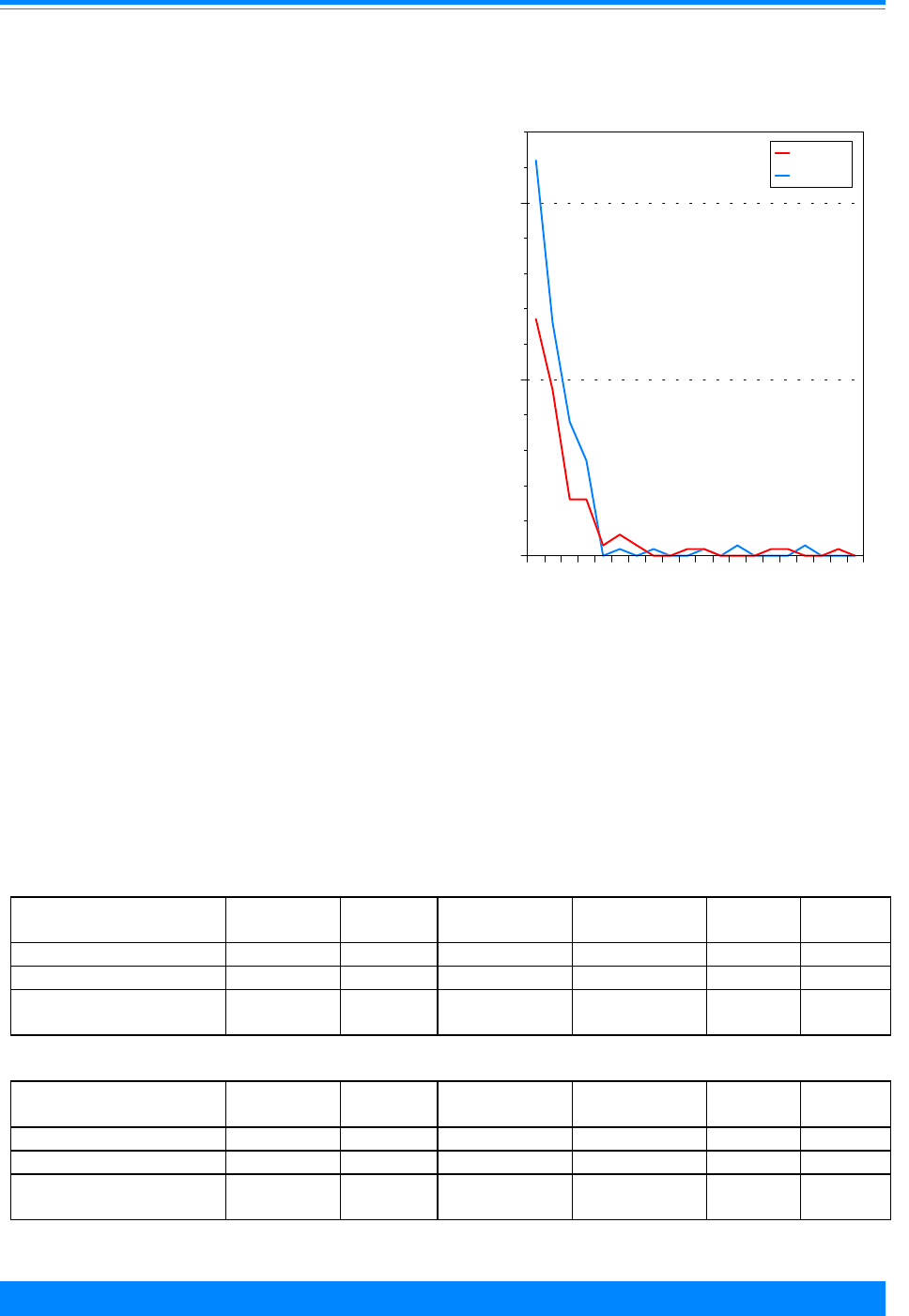
ICCC VII
93
National Cancer Institute SEER Pediatric Monograph
HEPATIC TUMORS
Sex and race-specific incidence
For children younger than 15 years of
age, the incidence of liver cancers was
slightly higher in males than females
(male:female ratio = 1.2) and somewhat
lower in black children compared with
white children (1.3 per million versus 1.6
per million). For children younger than 20
years of age, incidence rates were similar
for blacks and whites (1.4 per million
versus 1.5 per million) and were slightly
higher for males than females (male:female
= 1.2). The incidence of hepatoblastoma
was slightly higher in males than females
(male:female = 1.2), while the incidence of
hepatocellular carcinoma was similar in
both sexes (male:female = 1).
TRENDS
The incidence for total liver cancers in
children younger than 15 years of age
increased slightly from 1975 to 1995. The
rate was 1.4 per million for 1975-79 and
increased to 1.7 per million for 1990-95
(Table VII.1). For children younger than 20
years of age, there was little change in liver
cancer incidence during the 21-year period,
with rates between 1.4 and 1.6 per million
throughout the time period (Table VII.1).
The incidence of hepatoblastoma in-
creased markedly during the 1975-95
Figure VII.3: Hepatoblastoma and hepatocellular
carcinoma age-specific incidence rates
all races, both sexes
SEER, 1976-84, 1986-94
'
'
'
'
'
'
'
'
'
''
'
'
'
'''
'
''
+
+
+
++
+
+
+
++
++
+++
++
++
+
01234567891011121314151617181920
Age (in years) at diagnosis
0
5
10
Average annual rate per million
1976-84
1986-94
+
'
Table VII.1: Age-adjusted* incidence rates per million of liver cancers by age group, type,
and time period, all races, both sexes, SEER, 1975-95
<15 Years
Diagnosis ICCC
Category
1975-79 1980-84 1985-89 1990-95 1975-95
Hepatic tumors (total) VII(total) 1.4 1.6 1.7 1.7 1.6
Hepatoblastoma VIIa 0.8 1.1 1.4 1.5 1.3
Hepatocellular
carcinoma
VIIb 0.6 0.5 0.3 0.2 0.4
<20 Years
Diagnosis ICCC
Category
1975-79 1980-84 1985-89 1990-95 1975-95
Hepatic tumors (total) VII(total) 1.4 1.4 1.6 1.5 1.5
Hepatoblastoma VIIa 0.6 0.9 1.1 1.1 1.0
Hepatocellular
carcinoma
VIIb 0.7 0.6 0.5 0.4 0.5
*Adjusted to the 1970 US standard population

ICCC VII
94
National Cancer Institute
SEER Pediatric Monograph
HEPATIC TUMORS
period (Figure VII.4). The incidence rate
for children younger than 15 years of age
from 1975-79 was 0.8 per million and
increased to 1.5 per million for 1990-95
(Table VII.1). The incidence of hepatocellu-
lar carcinoma decreased during the period
1975-95, in contrast to the increase ob-
served for hepatoblastoma (Figure VII.4).
For children younger than 15 years of age,
the rate decreased from 0.6 per million in
1975-79 to 0.2 per million for 1990-95
(Table VII.1). Possible changes over time in
the assignment by histologic category could
only account for a small portion of the
observed opposite trends in incidence for
hepatoblastoma and hepatocellular carci-
noma.
SURVIVAL
Five-year survival rates for children
with hepatoblastoma improved from 51% to
59% between 1975-84 and 1985-94 (Figure
VII.5). Survival rates were substantially
lower for children and adolescents with
hepatocellular carcinoma, with an improve-
ment in 5-year survival rates from 31% for
the years 1975-84 to 42% for the years
1985-94 (Figure VII.5).
RISK FACTORS
Table VII.2 briefly summarizes current
knowledge on causes of hepatoblastoma.
The etiology of hepatoblastoma is as yet
unknown but there are some tantalizing
clues. One case-control study reported
elevated odds ratios with specific parental
occupational exposures, including maternal
exposures to metals, petroleum products,
and paints, and paternal exposures to
metals [2]. There have also been isolated
case reports of hepatoblastoma occurring in
association with fetal alcohol syndrome [3],
oral contraceptive use during pregnancy
[4], hormonal treatment for sterility [5],
and liver transplantation in the mother
combined with immunosuppressive treat-
ment throughout pregnancy [6]. Investiga-
Figure VII.4: Trends in liver cancer age-adjusted*
incidence rates by histology, age <20
all races, both sexes, SEER, 1975-95
#
#
#
#
#
##
#
#
#
#
#
#
#
#
#
#
#
#
#
#
'
'
''
''
'
'
'
'
''
'
'
'
''
'
'
'
'
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
1975 1980 1985 1990 1995
Year of diagnosis
0
0.5
1
1.5
2
2.5
3
Average annual rate per million
Total liver
Hepatoblastoma
Hepatocellular CA
"
'
#
*Adjusted to the 1970 US standard population
Figure VII.5: Hepatoblastoma and hepatocellular
carcinoma 5-year relative survival rates
age <20, all races, both sexes
SEER (9 areas), 1975-84 and 1985-94
51
31
59
42
Hepatoblastoma (VIIa) Hepatocellular CA (VIIb)
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94

ICCC VII
95
National Cancer Institute SEER Pediatric Monograph
HEPATIC TUMORS
tors in Japan recently noted that
hepatoblastoma accounted for more than
50% of early malignancies among Japanese
children who were of extremely low birth
weight (<1000gm) [7,8]. This finding raises
the possibility that factors associated with
prematurity and its treatment may play a
role in the occurrence of hepatoblastoma.
As a result, the marked improvement in
survival in recent years of extremely low
birth weight infants could in part be re-
sponsible for a notable increase in
hepatoblastoma rates in the United States
[9].
Hepatoblastoma has been associated
with familial adenomatous polyposis as
well as with syndromes involving
organomegaly (e.g., Beckwith-Wiedemann
syndrome and isolated hemihypertrophy)
[10,11]. Genes that are altered in the
tumor cells from some cases of
hepatoblastoma and that likely play an
important role in the pathogenesis of
hepatoblastoma include the APC gene
(which is responsible for familial
adenomatous polyposis) and the b-catenin
gene [12]. Another molecular abnormality
observed in some cases of hepatoblastoma
is loss of heterozygosity in the region of
chromosome 11 that is associated with
Beckwith-Wiedemann syndrome [13].
Hepatocellular carcinoma in children is
most common in regions of the world where
adult hepatocellular carcinoma is also
highly prevalent, for instance in sub-Sa-
haran Africa and Eastern Asia [1] and
among Alaskan Natives [14]. Chronic
infection with hepatitis B virus has been
implicated as the leading cause of hepato-
cellular carcinoma in children and young
adults. Universal hepatitis B immuniza-
tion will prevent the carrier state in chil-
dren and will lead to a dramatic reduction
in hepatocellular carcinoma, as demon-
strated during the past decade in Taiwan
[15]. Chronic infection with hepatitis C
virus (e.g., among hemophiliac males) is an
emerging risk factor for hepatocellular
carcinoma during adolescence [16]. Genes
that are altered in the tumor cells from
some cases of hepatocellular carcinoma and
that may play a role in its pathogenesis
include the β-catenin gene [17,18] and the
MET protooncogene [19].
Table VII.2: Current knowledge on causes of hepatoblastoma
Exposure or Characteristic Comments References
Beckwith-Wiedemann
syndrome, hemihypertrophy
Hepatoblastoma, Wilms’ tumor and adrenocortical
carcinoma are associated with these syndromes that
involve organomegaly.
11,20,21
Family history of familial
adenomatous polyposis and
Gardner’s syndrome
Both these syndromes involve multiple colonic
polyps, have an autosomal dominant inheritance,
and are caused by mutations in the APC gene.
10,22-24
Parental occupational
exposures
Associations with metals, petroleum products, paints
and pigments were reported from the only case-
control study done to date.
2
Known risk factors
Factors for which evidence is
inconsistent or limited

ICCC VII
96
National Cancer Institute
SEER Pediatric Monograph
HEPATIC TUMORS
SUMMARY
Liver cancers are uncommon in chil-
dren and represented only 1.1% of malig-
nancies in SEER areas for children younger
than 20 years of age, with an annual inci-
dence rate of 1.5 per million (1975-95).
Hepatoblastoma was the most common
malignancy of the liver in children and its
incidence was highest during the first year
of life and decreased rapidly with increas-
ing age. Hepatocellular carcinoma was the
second most common malignancy of the
liver and occurred primarily among adoles-
cents. While the incidence of
hepatoblastoma increased from 1975-95,
the incidence of hepatocellular carcinoma
decreased.
Reference List
1. Parkin DM, Stiller CA, Draper GJ, et al: The
international incidence of childhood cancer. Int
J Cancer 42:511-520, 1988.
2. Buckley JD, Sather H, Ruccione K, et al: A
case-control study of risk factors for
hepatoblastoma. A report from the Childrens
Cancer Study Group. Cancer 64:1169-76, 1989.
3. Khan A, Bader JL, Hoy GR, et al:
Hepatoblastoma in child with fetal alcohol
syndrome [letter]. Lancet 1:1403-4, 1979.
4. Otten J, Smets R, De Jager R, et al:
Hepatoblastoma in an infant after contracep-
tive intake during pregnancey [letter]. N Engl
J Med 297:222, 1977.
5. Melamed I, Bujanover Y, Hammer J, et al:
Hepatoblastoma in an infant born to a mother
after hormonal treatment for sterility [letter].
N Engl J Med 307:820, 1982.
6. Roll C, Luboldt HJ, Winter A, et al:
Hepatoblastoma in a 2-year-old child of a liver-
transplanted mother. Lancet 349:103, 1997.
7. Ikeda H, Matsuyama S, Tanimura M: Associa-
tion between hepatoblastoma and very low
birth weight: a trend or a chance? [see com-
ments]. J Pediatr 130:557-60, 1997.
8. Tanimura M, Matsui I, Abe J, et al: Increased
risk of hepatoblastoma among immature
children with a lower birth weight. Cancer Res
58:3032-5, 1998.
9. Ross JA, Gurney JG: Hepatoblastoma inci-
dence in the United States from 1973 to 1992.
Med Pediatr Oncol 30:141-2, 1998.
10. Giardiello FM, Offerhaus GJ, Krush AJ, et al:
Risk of hepatoblastoma in familial
adenomatous polyposis. J Pediatr 119:766-8,
1991.
11. DeBaun MR, Tucker MA: Risk of cancer during
the first four years of life in children from The
Beckwith-Wiedemann Syndrome Registry [see
comments]. J Pediatr 132:398-400, 1998.
12. Koch A, Denkhaus D, Albrecht S, et al: Child-
hood hepatoblastomas frequently carry a
mutated degradation targeting box of the beta-
catenin gene. Cancer Res 59:269-73, 1999.
13. Koufos A, Hansen M, Copeland N, et al: Loss of
heterozygosity in three embryonal tumours
sugests a common pathogenic mechanism.
Nature 316:330-334, 1985.
14. Lanier AP, McMahon BJ, Alberts SR, et al:
Primary liver cancer in Alaskan natives. 1980-
1985. Cancer 60:1915-20, 1987.
15. Chang MH, Chen CJ, Lai MS, et al: Universal
hepatitis B vaccination in Taiwan and the
incidence of hepatocellular carcinoma in
children. Taiwan Childhood Hepatoma Study
Group [see comments]. N Engl J Med
336:1855-9, 1997.
16. Darby SC, Ewart DW, Giangrande PL, et al:
Mortality from liver cancer and liver disease in
haemophilic men and boys in UK given blood
products contaminated with hepatitis C. UK
Haemophilia Centre Directors’ Organisation.
Lancet 350:1425-31, 1997.
17. de La Coste A, Romagnolo B, Billuart P, et al:
Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular
carcinomas. Proc Natl Acad Sci U S A 95:8847-
51, 1998.
18. Miyoshi Y, Iwao K, Nagasawa Y, et al: Activa-
tion of the beta-catenin gene in primary
hepatocellular carcinomas by somatic alter-
ations involving exon 3. Cancer Res 58:2524-7,
1998.
19. Park WS, Dong SM, Kim SY, et al: Somatic
mutations in the kinase domain of the Met/
hepatocyte growth factor receptor gene in
childhood hepatocellular carcinomas. Cancer
Res 59:307-10, 1999.
20. Ishak KG, Glunz PR: Hepatoblastoma and
hepatocarcinoma in infancy and childhood.
Report of 47 cases. Cancer 20:396-422, 1967.
21. Fraumeni JF, Jr., Miller RW, Hill JA: Primary
carcinoma of the liver in childhood: an epide-
miologic study. J Natl Cancer Inst 40:1087-99,
1968.
22. Kingston JE, Herbert A, Draper GJ, et al:
Association between hepatoblastoma and
polyposis coli. Arch Dis Child 58:959-62, 1983.

ICCC VII
97
National Cancer Institute SEER Pediatric Monograph
HEPATIC TUMORS
23. Garber JE, Li FP, Kingston JE, et al:
Hepatoblastoma and familial adenomatous
polyposis [published erratum appears in J Natl
Cancer Inst 1989 Mar 15;81(6):461]. J Natl
Cancer Inst 80:1626-8, 1988.
24. Krush AJ, Traboulsi EI, Offerhaus JA, et al:
Hepatoblastoma, pigmented ocular fundus
lesions and jaw lesions in Gardner syndrome.
Am J Med Genet 29:323-32, 1988.

98
National Cancer Institute
SEER Pediatric Monograph

ICCC VIIIMALIGNANT BONE TUMORS
99
National Cancer Institute SEER Pediatric Monograph
HIGHLIGHTS
Incidence
♦ Malignancies of the bone, with an average annual incidence rate of 8.7 per million
children younger than 20 years of age, comprised about 6% of childhood cancer
reported by SEER areas from 1975-95.
♦ In the US, 650-700 children and adolescents younger than 20 years of age are
diagnosed with bone tumors each year of which approximately 400 are osteosar-
coma and 200 are Ewing’s sarcoma.
♦ The two types of malignant bone cancer that predominated in children were os-
teosarcomas and Ewing’s sarcomas, about 56% and 34% of the malignant bone
tumors, respectively.
♦ Osteosarcomas derive from primitive bone-forming mesenchymal stem cells and
most often occur near the metaphyseal portions of the long bones. The Ewing’s
sarcomas are believed to be of neural crest origin and occur roughly evenly between
the extremities and the central axis.
♦ For all bone cancer combined, a steady rise in incidence rates occurred with increas-
ing age between ages 5 and 10, and a steeper rise began at age 11 until age 15
coinciding with the adolescent growth spurt. The peak incidence of bone cancer (19
per million) occurred at age 15, after which rates showed a decline (Figure VIII.2).
♦ Rates did not differ much by sex among younger children, but males had higher
incidence than females during adolescence (Figure VIII.4).
♦ For osteosarcoma, black children had a higher overall rate than did white children
(Figure VIII.7). For Ewing’s sarcoma the racial variation in rates was dramatic:
white children had an approximate 6-fold higher incidence rate than black children
(Figure VIII.8).
♦ The most frequent site of bone cancer development was the long bones of the lower
limbs for osteosarcomas and the central axis for Ewing’s sarcomas (Figure VIII.9).
Survival
♦ The 5-year relative survival for children with bone cancer improved from 49% in the
period 1975-84, to 63% in the period 1985-94. The survival rates improved between
the two time periods for both osteosarcoma (Figure VIII.11) and Ewing’s sarcoma
(Figure VIII.12).
♦ Survival rates for osteosarcoma were higher than those for Ewing’s sarcoma espe-
cially in the earlier time period (Figures VIII.11 and VIII.12).
Risk factors
♦ Although directed ionizing radiation exposure and a few genetic susceptibility
syndromes are associated with increased risk of osteosarcoma, to date no factor has
emerged to explain even a modest proportion of cases (Table VIII.2). Other than
the important racial difference in incidence between black and white children, no
environmental factor or other characteristic has yet been shown to be a strong risk
factor for Ewing’s sarcoma (Table VIII.3).
James G. Gurney, Andrine R. Swensen, Marc Bulterys

ICCC VIII
MALIGNANT BONE TUMORS
100
National Cancer Institute
SEER Pediatric Monograph
INTRODUCTION
This chapter describes the descriptive
epidemiology of childhood bone cancer,
including short discussions on survival and
risk factors for occurrence. Sarcomas of the
bone and cartilage are a diverse group of
tumors comprising about 0.5% of all malig-
nancies in humans. The relative magni-
tude of bone cancer, however, is consider-
ably higher in children than in adults [1].
About half of bone tumors that occur
among children are of nonmalignant histo-
pathology [2]. Because SEER case report-
ing is limited to primary malignant neo-
plasms, the information presented in this
report will refer only to malignancies of the
bone (bone cancer). In the International
Classification of Childhood Cancer (ICCC)
classification system, bone cancers are
categorized as osteosarcomas, Ewing’s
sarcomas, chondrosarcomas, ‘other specified
malignant bone tumors’ and ‘unspecified
malignant bone tumors’ [3]. The two types
of bone cancer that predominate in children
are osteosarcomas and Ewing’s sarcomas.
For the 21-year period of 1975-95, there
were 1,657 children younger than 20 years
of age in the SEER areas who were diag-
nosed with a primary bone malignancy.
Osteosarcomas represented about 56% of
these tumors and Ewing’s sarcomas an
additional 34%. In the US, 650-700 chil-
dren and adolescents younger than 20
years of age are diagnosed with bone
tumors each year of which approximately
400 are osteosarcoma and 200 are Ewing’s
sarcoma.
Osteosarcomas derive from primitive
bone-forming mesenchymal stem cells and
most often occur near the metaphyseal
portions of the long bones [3]. There is a
bimodal age distribution of osteosarcoma
incidence, with peaks in early adolescence
and in adults older than 65 years of age [1].
The Ewing’s sarcomas, which include
Ewing’s, atypical Ewing’s, and the periph-
eral primitive neuroectodermal tumor of
bone, are believed to be of neural crest
Figure VIII.1: Percent distribution of bone cancers by histology
and age group, all races, both sexes, SEER, 1975-95
53.9
37
4.2
4.9
55.5
33.7
6
4.8
Osteosarcoma
Ewing's sarcoma
Chondrosarcoma
Others
010203040506070 0 10203040506070
Age <15 years
Age <20 years
Relative percent

ICCC VIIIMALIGNANT BONE TUMORS
101
National Cancer Institute SEER Pediatric Monograph
origin and occur roughly evenly between
the extremities and the central axis [5].
Ewing’s sarcoma is a disease primarily of
childhood and young adults; occurrence in
older adults is extremely rare [1]. Chondro-
sarcomas, which after osteosarcomas are
the most common of the bone malignancies
among adults [1], are very rare in children.
Figure VIII.1 presents the relative distribu-
tion of bone cancer by histologic types, both
for children younger than 15 years of age
and younger than 20 years of age.
INCIDENCE
Malignancies of the bone, with an
average annual incidence rate of 8.7 per
than 20 years, unless otherwise noted).
The histology-specific rates were 4.8 per
million for osteosarcoma, 2.9 per million for
Ewing’s sarcoma and 0.5 per million for
chondrosarcoma.
Age-specific incidence
Bone cancer represented only 0.5% of
all malignancies among children younger
than 5 years, compared with 5% for those
5-9 years, 11% for those 10-14 years, and
8% for adolescents 15-19 years. Figure
VIII.2 shows 1-year age-specific rates for
all bone cancer combined and for specific
histologic subtypes.
1
For all bone cancer
combined, a steady rise in rates occurred
from ages 5 through 10, and a steeper rise
began at age 11. The increase in rates
among older children appeared to coincide
with the adolescent growth spurt. The
peak incidence of bone cancer (19 per
million) occurred at age 15, after which
rates showed a decline. Incidence of chond-
Figure VIII.2: Bone cancer age-specific incidence rates by histology
all races, both sexes, SEER, 1976-84 and 1986-94 combined
&
&
&
&&&&&&
&
&
&
&&
&&
&
&
&
&
(
(
(((
((
(
(
(
(
(
(
(
(
(
(
(
(
(
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
All Bone
Osteosarcoma
Ewing's Sarcoma
Chondrosarcoma
#
,
(
&
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.
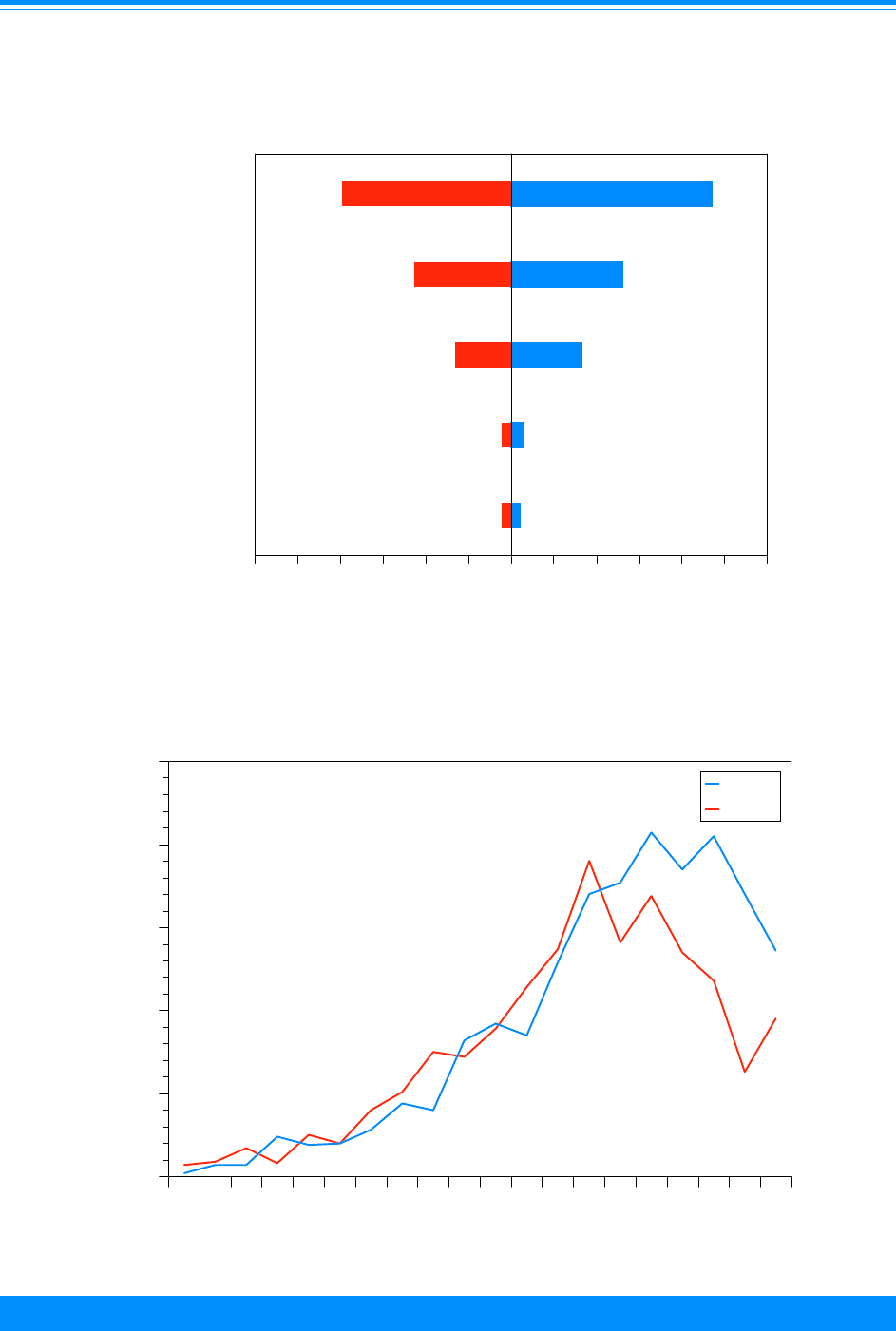
ICCC VIII
MALIGNANT BONE TUMORS
102
National Cancer Institute
SEER Pediatric Monograph
*Adjusted to the 1970 US standard population
9.4
5.2
3.3
0.6
0.4
7.9
4.5
2.6
0.4
0.4
All Bone
Osteosarcoma
Ewing's sarcoma
Chondrosarcoma
Others
Histology
024681012 024681012
Female
Male
Average annual rate per million
Figure VIII.3: Bone cancer age-adjusted incidence* rates
by type and sex, age <20, all races, SEER, 1975-95
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
""
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
01234567891011121314151617181920
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
Male
Female
"
)
Figure VIII.4: Bone cancer age-specific incidence rates
by sex, all races, SEER, 1976-84 and 1986-94

ICCC VIIIMALIGNANT BONE TUMORS
103
National Cancer Institute SEER Pediatric Monograph
rosarcoma was very low at all ages. Rates
of osteosarcoma and Ewing’s sarcoma were
similar until about age 10, when substan-
tially higher rates of osteosarcoma became
apparent.
Sex-specific incidence
The incidence rates of osteosarcoma
and Ewing’s sarcoma were slightly higher
for males relative to females, albeit the
absolute differences in rates were quite
small (Figure VIII.3). Figure VIII.4 pre-
sents 1-year age and sex specific incidence
rates for all bone cancer combined. The
incidence pattern by age is similar for
males and females, although from age 14
through 19 male rates are higher than
female rates. For females, rates of bone
cancer peaked at age 13, while the highest
rates for males occurred from ages 15
through 17.
Black-white differences in incidence
One-year age specific incidence rates of
bone cancer are shown in FigureVIII.5 for
white and black children. The age pattern
of bone cancer incidence was quite similar
by race, although higher rates among
whites were seen at virtually all ages. The
overall incidence rate among white children
was 8.8 per million compared with 6.8 per
million for black children. Figure VIII.6
shows that both white males and females
had higher rates than blacks of the same
sex, at about the same ratios. This racial
disparity in bone cancer incidence was not
consistent across histologic subtypes. For
osteosarcoma, black children had a higher
overall rate than did white children (Figure
VIII.7). Rates were slightly higher in
blacks than in whites for each age group
except for those younger than 5 years of
age. For Ewing’s sarcoma the racial varia-
Figure VIII.6: Bone cancer age-adjusted* incidence rates
by race and sex, age <20, SEER, 1975-95
*Adjusted to the 1970 US standard population
Average annual rate per million
9.6
7.5
9.4
8.1
6.2
7.9
Whites
Blacks
All Races
Race
024681012 0 2 4 6 8 10 12
Female
Male
Figure VIII.5: Bone cancer age-specific incidence rates
by race, both sexes, SEER, 1976-84 and 1986-94 combined
'
'
''
''
'
'
'
'
'
'
'
'
'
'
'
'
'
'
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
5
10
15
20
25
Average annual rate per million
White
Black
+
'

ICCC VIII
MALIGNANT BONE TUMORS
104
National Cancer Institute
SEER Pediatric Monograph
Figure VIII.7: Osteosarcoma age-adjusted* incidence rates
by age group and race, both sexes, SEER, 1975-95
*Adjusted to the 1970 US standard population
0.5
2.1
7
8.2
4.6
0.3
2.6
8.3
8.9
5.2
<5
5 - 9
10 - 14
15 - 19
<20
Age (in years) at diagnosis
024681012 024681012
Black
White
Average annual rate per million
Figure VIII.8: Ewing's sarcoma age-adjusted* incidence rates
by age group and race, both sexes, SEER, 1975-95
*Adjusted to the 1970 US standard population
0.6
2.5
5
5.1
3.4
0.3
0.5
0.5
0.9
0.6
<5
5 - 9
10 - 14
15 - 19
<20
Age (in years) at diagnosis
0246810 0246810
Black
White
Average annual rate per million
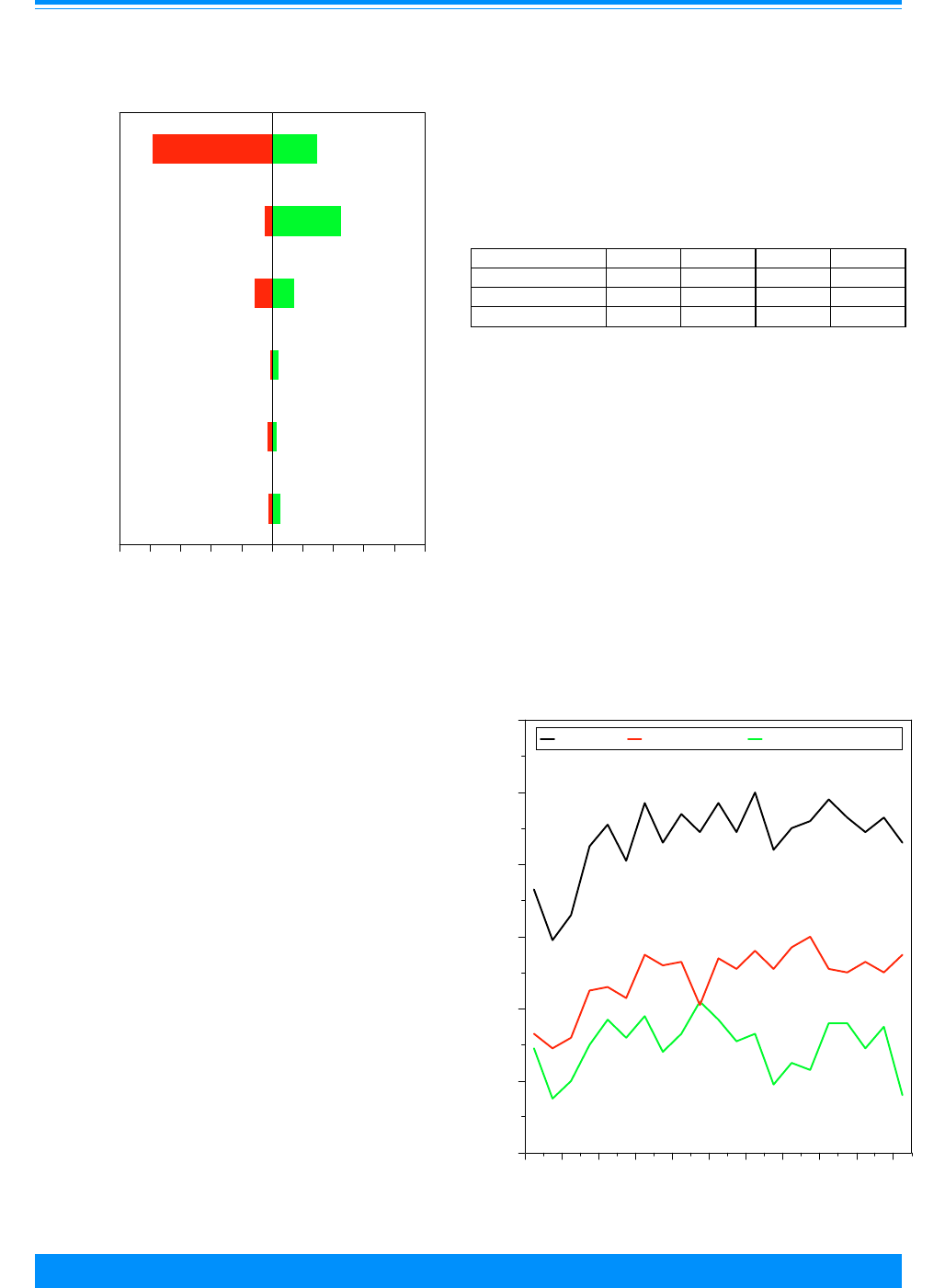
ICCC VIIIMALIGNANT BONE TUMORS
105
National Cancer Institute SEER Pediatric Monograph
tion in rates was dramatic: white children
had an approximate 6-fold higher incidence
rate than black children (Figure VIII.8),
thus entirely accounting for the white
preponderance in overall bone cancer rates.
This strong racial difference was apparent
in all age groups. The fact that black
children in the US rarely develop Ewing’s
sarcoma has been observed for many years,
but the protective etiology has yet to be
elucidated. It is interesting to note that in
several African countries the ratio of
Ewing’s sarcoma to osteosarcoma is very
similar to that of US blacks [6].
Bone cancer location
The most frequent site of bone cancer
development (57%) was the long bones of
the lower limbs. The site distribution of
Ewing’s sarcomas, however, differed sub-
stantially from that of osteosarcomas
(Figure VIII.9). The long bones of the lower
limb were the site of 78% of osteosarcomas,
Figure VIII.10: Trends in bone cancer age-adjusted*
incidence rates by histology, age <20 all races
both sexes, SEER, 1975-95
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
((
(
(
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
75 77 79 81 83 85 87 89 91 93 95
Year of diagnosis
0
2
4
6
8
10
12
Average annual rate per million
All bone Ostesarcoma Ewing's sarcoma
# , (
*Adjusted to the 1970 US standard population
Figure VIII.9: Anatomic site distribution of bone
cancer by histology, age <20, all races, both sexes
SEER, 1975-95
78.2
4.9
11.3
1
2.7
1.9
29.4
45
14.2
3.8
2.7
4.9
Long bones/
Central axis
Long bones/
Short bones/
Face or skull
All other sites
Anatomic site
020406080100 0 20 40 60 80 100
Lower limbs
Upper or lower
Upper limbs
Osteosarcoma
Ewing's sarcoma
Relative percent
but only 29% of Ewing’s sarcomas. The
central axis (vertebral column; rib, ster-
num, and clavicle; pelvic, sacrum, and
coccyx) was the most frequent site for
Ewing’s sarcomas (45%), where osteosarco-
mas are relatively unusual.
Trends in incidence rates
Figure VIII.10 shows histology-specific
incidence rates by single year of diagnosis
from 1975-95. It is unclear why rates of
both osteosarcoma and Ewing’s sarcoma
were lower from 1975-78 than in later
years. Table VIII.1 shows the average
rates of bone cancer during the time peri-
ods of this study.
Table VIII.1: Average age-adjusted* incidence rates
per million children for bone cancer
all races, both sexes, age<20, SEER 1975-95
1975-79 1980-84 1985-89 1990-95
Osteosarcoma 3.7 4.9 5.4 5.3
Ewing’s Sarcoma 2.6 3.4 2.9 2.9
All Bone Cancer 7.4 9.0 9.2 9.2
*Adjusted to the 1970 US standard population
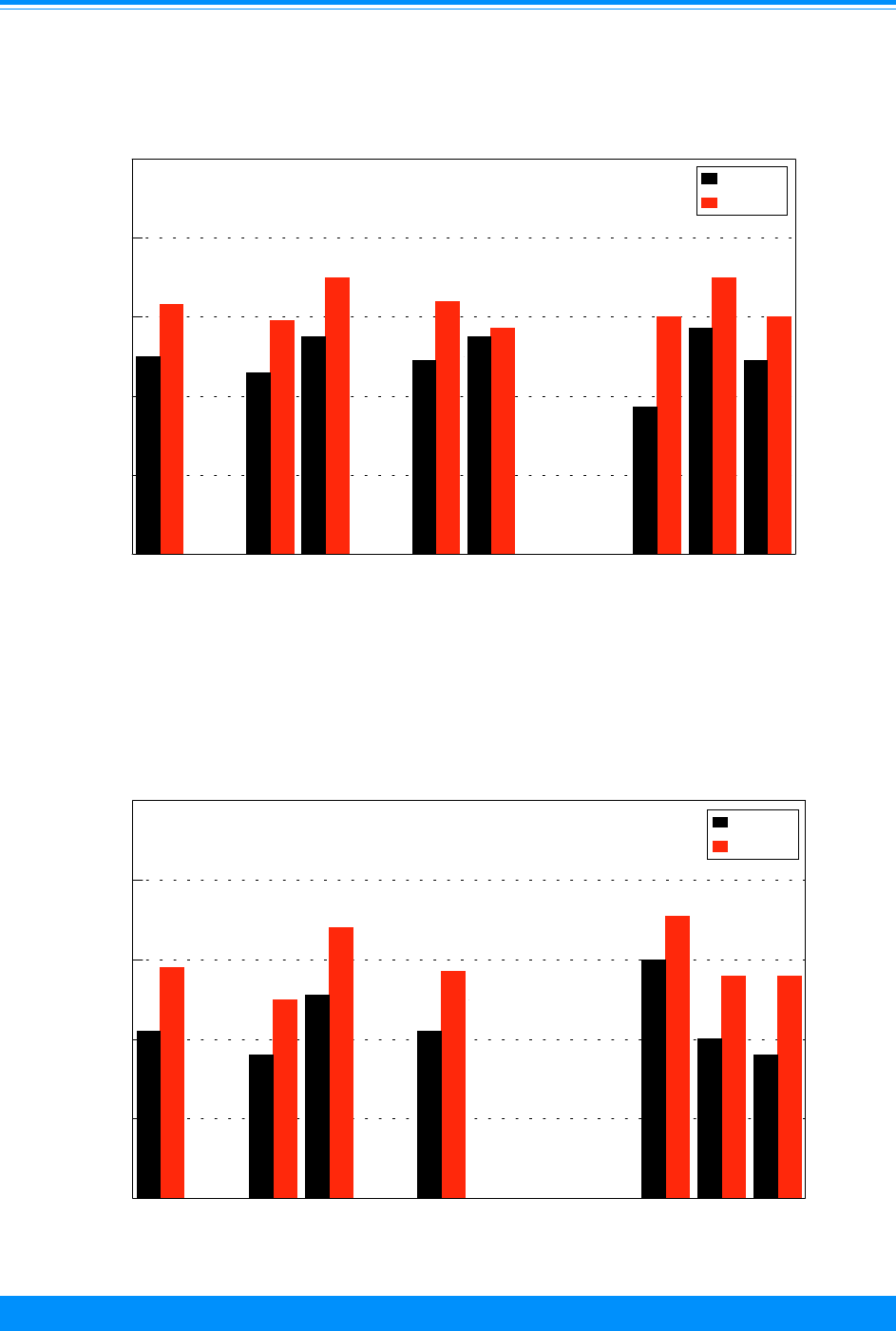
ICCC VIII
MALIGNANT BONE TUMORS
106
National Cancer Institute
SEER Pediatric Monograph
Figure VIII.11: Osteosarcoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
50
46
55
49
55
37
57
49
63
59
70
64
57
60
70
60
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race Age
# - <25 cases - rate not shown
#
Figure VIII.12: Ewing's sarcoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
42
36
51
42
60
40
36
58
50
68
57
71
56 56
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
##
# - <25 cases - rate not shown
Sex Race Age

ICCC VIIIMALIGNANT BONE TUMORS
107
National Cancer Institute SEER Pediatric Monograph
SURVIVAL
The 5-year relative survival for
children with bone cancer improved from
49% in the period 1975-84, to 63% in the
period 1985-94. The time period for
relative survival is 1985-94 unless other-
wise noted. Females had better 5-year
survival probability (70%) than males
(59%) and there was only a slight differ-
ence in survival between blacks (60%)
and whites (63%). No notable survival
differences were observed across 5-year
age groups. For osteosarcoma the 5-year
relative survival was 63% (59% for males,
70% for females). Prognosis for Ewing’s
sarcoma was somewhat poorer than for
ostoesarcoma. The overall 5-year relative
survival for Ewing’s sarcoma was 58%, and
again there was a notable difference by sex
(50% for males, 68% for females). Although
survival did not differ substantially by tumor
site for osteosarcoma, children with Ewing’s
sarcoma of the pelvic, sacrum, or coccyx has
survival probabilities of under 35%.
Known risk factors
Factors for which
evidence is limited or
inconsistent
Table VIII.2: Current knowledge on causes of osteosarcoma
Exposure or Characteristic Comments References
Prior treatment for childhood
cancer with radiation
therapy and/or chemotherapy
There is an increased risk following radiotherapy
for childhood cancer.
Independent of radiotherapy, treatment with
alkylating agents increases the risk of developing
osteosarcoma.
7-9
Hereditary retinoblastoma,
Li-Fraumeni syndrome, and
Rothmund-Thomson
syndrome
Increased risk is well documented for these
genetic conditions.
10-13
Radium High doses of the radioisotope radium are known
to cause osteosarcoma in adults. Whether the low
levels sometimes found in drinking water confer
risk to children or adults is unknown.
1,14
Growth and development There has been some suggestion that taller
stature is associated with an increased risk, but
the results of more recent studies do not support
this finding. One study showed an association
with earlier age at onset of secondary sex
characteristics in females and lower weight gain
during pubertal growth spurt in males.
15-19
Prior trauma to tumor site One study found a small positive association
between damage to the tumor site and increased
risk of osteosarcoma.
16
Prenatal exposure and
development
Short birth length and fetal x-rays were
associated with an increased risk in a single
study.
16
Parental exposures An association with chicken farming and another
with gardening with fertilizer, herbicides or
pesticides have been reported in single studies.
20-21
Fluoride in drinking water The few epidemiologic studies as well as ecologic
and time trend analyses suggest that fluoride is
unlikely to cause osteosarcoma.
22-25

ICCC VIII
MALIGNANT BONE TUMORS
108
National Cancer Institute
SEER Pediatric Monograph
RISK FACTORS
Unfortunately, the current state of
knowledge regarding the causes of bone
cancer is limited. Table VIII.2 briefly
summarizes results from a number of
epidemiologic studies that have been con-
ducted on children with osteosarcoma.
Although directed ionizing radiation expo-
sure and a few genetic susceptibility syn-
dromes are associated with increased risk
of osteosarcoma, to date no factor has
emerged to explain even a modest propor-
tion of cases. The same is true for Ewing’s
sarcoma. Other than the important racial
difference in incidence between black and
white children, no environmental factor or
other characteristic has yet been shown to
be a strong risk factor for Ewing’s sarcoma
(TableVIII.3).
SUMMARY
In this descriptive analysis of the
population-based SEER data, bone cancer
represented about 6% of malignancies in
children younger than age 20 years, with
an average annual incidence rate of 8.7
cases per million children from 1975-95 (9.2
per million from 1990-95). Incidence in-
creased with increasing age until late
adolescence. Rates did not differ much by
sex among younger children, but males had
higher incidence than females during
adolescence. Osteosarcoma and Ewing’s
sarcomas were the most common malignan-
cies of bone in children. Black children had
slightly higher rates of osteosarcoma rela-
tive to white children, while incidence of
Ewing’s sarcoma was dramatically higher
among white compared with black children.
The most common site for development of
Risk factors for which
evidence is limited or
inconsistent
Table VIII.3: Current knowledge on causes of Ewing’s Sarcoma (ES)
Exposure or Characteristic Comments References
Race ES is almost exclusively a disease of white children.
Rates in whites are approximately 9 times those in blacks.
18,26,27
Growth As for osteosarcoma, recent studies have not found a
consistent association with increased height or weight, or
age at pubertal growth spurt.
15,18,27-30
Hernia An association was found between hernias and increased
risk in one study.
29
Paternal occupation Paternal occupation in agriculture has been associated
with increased risk in two studies, although only in one
were the results statistically significant.
29,30
Ingestion of poison or
overdose of medication
A prior poisoning episode was more common among cases
than controls in a single study.
30
Family history of cancer ES has been reported in several pairs of siblings.
However, more than one family member with ES is rare.
In a study of over 200 cases, none had a relative with ES.
Unlike osteosarcoma, ES is not part of the Li-Fraumeni
syndrome.
12,31-32
Known risk factors

ICCC VIIIMALIGNANT BONE TUMORS
109
National Cancer Institute SEER Pediatric Monograph
Reference List
1. Miller RW, Boice JD Jr, Curtis RE. Bone Cancer.
Schottenfeld D, Fraumeni JF, Editors. Cancer
Epidemiology and Prevention. 2nd ed. New
York: Oxford University Press; 1996;44:971-983.
2. Dahlin DC, Unni KK. Bone Tumors: General
Aspects and Data on 8542 Cases. 4th ed.
Springfield, IL: Charles C. Thomas; 1986.
3. Kramarova E, Stiller CA. The international
classification of childhood cancer. Int J Cancer.
1996;68:759-765.
4. Link MP, Eiler F. Osteosarcoma. Pizzo PA,
Poplack DG, Editors. Principles and Practices of
Pediatric Oncology. 3rd ed. Philadelphia, PA:
Lippencott-Raven; 1997:889-919.
5. Horowitz ME, Malawer MM, Woo SY, Hicks MJ.
Ewing’s Sarcoma Family of Tumors: Ewing’s
Sarcoma of Bone and Soft Tissue and the
Peripheral Primitive Neuroectodermal Tumors.
Pizzo PA, Poplack DG, Editors. Principles and
Practices of Pediatric Oncology. 3rd ed. Philadel-
phia, PA: Lippencott-Raven; 1997:831-888.
6. Parkin DM, Stiller CA, Draper GJ, Bieber CA.
The international incidence of childhood cancer.
Int J Cancer. 1988;42:511-520.
7. Tucker MA, D’Angio GJ, Boice JD,et al. Bone
sarcomas linked to radiotherapy and chemo-
therapy in children. New Eng J Med
1987;317:588-593.
8. Hawkins MM, Wilson LMK, Burton HS, et al.
Radiotherapy, alkylating agents, and risk of
bone cancer after childhood cancer. J Natl
Cancer Inst. 1996;88:270-278.
9. Newton WA, Meadows AT, Shimada H, et al.
Bone sarcomas as a second malignant neoplasm
following childhood cancer. Cancer 1991;67:193-
201.
10. Wong Fl, Boice JD, Abramson DH, et al. Cancer
incidence after retinoblastoma. Radiation dose
and sarcoma risk. JAMA 1997;278:1262-1267.
11. Hansen MF, Koufos A, Gallie BL, et al. Osteosa-
rcoma and retinoblastoma: a shared chromo-
somal mechanism revealing recessive predispo-
sition. Proc Natl Acad Sci 1985;82:6216-6220.
12. Li FP, Fraumeni JF, Mulvihill JJ, et al. A cancer
family syndrome in twenty-four kindreds.
Cancer Res 1988; 48:5358-5362.
13. Leonard A, Craft AW, Moss C, and Malcolm AJ.
Osteogenic sarcoma in the Rothmund-Thomson
syndrome. Med Pediatr Oncol 1996;26:249-253.
14. Finkelstein MM and Kreiger N. Radium in
drinking water and risk of bone cancer in
Ontario youths: a second study and combined
analysis. Occup Environ Med. 1996 May;
53(5):305-11.
15. Fraumeni JF, Jr. Stature and malignant tumors
of bone in childhood and adolescence. Cancer
1967;20:967-973.
16. Operskalski EA, Preston-Martin S, Henderson
BE, and Visscher BR. A case-control study of
osteosarcoma in young persons. Am J Epidemiol
1987;126:118-126.
17. Pui CH, Dodge RK, George SL, and Green AA.
Height at diagnosis of malignancies. Arch Dis
Child 1987;62:495-499.
18. Buckley JD, Pendergrass TW, Buckley CM,
Pritchard DJ, Nesbit ME, Provisor AJ and
Robison LL. Epidemiology of osteosarcoma and
Ewing’s sarcoma in childhood: A study of 305
cases from the Children’s Cancer Group. Cancer.
Cancer 1998;83:1440-8.
19. Gelberg KH, Fitzgerald EF, Hwang SA, and
Dubrow R. Growth and development and other
risk factors for osteosarcoma in children and
adults. In J Epidemiol 1997;26:272-278.
20. Schwartzbaum JA, George SL, Pratt CB and
Davis B. An exploratory study of environmental
and medical factors potentially related to
childhood cancer. Med Pediatr Oncol.
1991;19:115-121.
21. Kristensen P, Andersen A, Irgens LM, Bye AS,
Sundheim L. Cancer in offspring of parents
engaged in agricultural activities in Norway:
Incidence and risk factors in the farm environ-
ment. Int J Cancer 1996; 65:39-50.
22. Moss ME, Kanarek MS, Anderson HA, et al.
Osteosarcoma, seasonality and environmental
factors in Wisconsin, 1979-1989. Arch Environ
Health 1995;50:235-241.
23. Young FE. Public health report on fluoride
benefits and risks. JAMA 1991; 266:1061-1067.
osteosarcoma was the long bones of the
lower limbs, while Ewing’s sarcoma most
frequently developed in bones of the central
axis. Except for the first few years of the
data collection, incidence rates of bone
cancer have been stable. The etiology of
bone cancer remains uncertain and the few
risk factors that have been identified
explain only a very small proportion of the
incidence of these diseases. The 5-year
relative survival for children with bone
cancer improved from 49% in the period
1975-84, to 63% in the period 1985-94. In
general, 5-year relative survival for os-
teosarcoma was slightly better than for
Ewing’s sarcoma. For both diseases, how-
ever, females had notably better survival
than males.

ICCC VIII
MALIGNANT BONE TUMORS
110
National Cancer Institute
SEER Pediatric Monograph
24. McGuire SV, Vanable ED, McGuire MH, et al. Is
there a link between fluoridated water and
osteosarcoma? J Am Dent Assoc. 1991;122:38-
45.
25. Gelberg KH, Fitzgerald EF, Hwang SA, and
Dubrow R. Fluoride exposure and childhood
osteosarcoma: a case-control study. Am J Pub
Health 1995;85:1678-1683.
26. Gurney JG, Severson RK, Davis S, and Robison
LL. Incidence of cancer in children in the United
States. Cancer 1995;75:2186-2195.
27. Polednak AP. Primary bone cancer incidence in
black and white residents of New York state.
Cancer 1985;55:2883-2888.
28. Pendergrass TW, Foulkes MA, Robison LL, and
Nesbit ME. Stature and Ewing’s sarcoma in
childhood. Am J Pediatr Hematol Oncol
1984;6:33-39.
29. Winn DM, Li FP, Robison LL, Mulvihill JJ, and
Fraumeni JF, Jr. A case-control study of the
etiology of Ewing’s sarcoma. Cancer Epidemiol
Biomarkers Prev 1992;1:525-532.
30. Holly EA, Aston DA, Ahn DK, and Kristiansen
JJ. Ewing’s bone sarcoma, paternal occupational
exposure, and other factors. Am J Epidemiol
1992; 135:122-129.
31. Hartley AL, Birch JM, Blair CV, Teare MD,
Marsden HB, and Harris M. Cancer Incidence
in the families of children with Ewing’s sar-
coma. J Natl Cancer Inst. 1991 Jul 3;83(13):955-
6.
32. Novakovic B, Tucker MA, Wexier L, Horowitz M,
McLure L. Risk of cancer in families of patients
with Ewing’s sarcoma family of tumors. Ann
Meet Am Assoc Cancer Res 1994;35:A1729.

ICCC IX
111
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
James G. Gurney, John L. Young, Jr., Steven D. Roffers, Malcolm A. Smith, Greta R. Bunin
HIGHLIGHTS
Incidence
♦ The soft tissue sarcomas of children and adolescents arise primarily from the con-
nective tissues of the body, such as fibrous tissue, adipose tissue, and muscle tissue.
The sarcomas that arise from bone are discussed separately in the bone tumor
chapter.
♦ In the US, 850-900 children and adolescents younger than 20 years of age are diag-
nosed with soft tissue sarcomas each year, of which approximately 350 are rhab-
domyosarcomas.
♦ The incidence of soft tissue sarcomas for children and adolescents younger than 20
years of age was 11.0 per million (Table IX.2), representing 7.4% of cancer cases for
this age group.
♦ Rhabdomyosarcoma was the most common soft tissue sarcoma among children 0-14
years, representing nearly 50% of soft tissue sarcomas for this age range (Figure
IX.1) with an incidence rate of 4.6 per million (Table IX.2).
♦ There are two major types of rhabdomyosarcoma: embryonal (about 75% of rhab-
domyosarcoma cases) and alveolar. These two subtypes tended to occur at different
body sites (Figure IX.3) and had different age patterns (Figure IX.2). The incidence
of embryonal rhabdomyosarcoma was higher among children 0-4 years, while the
incidence of alveolar rhabdomyosarcoma was similar throughout childhood (Figure
IX.2).
♦ Other types of soft tissue sarcomas are rare and the incidence is higher in adoles-
cents compared to younger children. Among these are the fibrosarcomas, malignant
fibrous histiocytoma, synovial sarcoma, leiomyosarcoma, liposarcoma, and others
(Table IX.2).
♦ For infants, the most common soft tissue sarcoma was embryonal rhabdomyosar-
coma. However, a distinctive set of other soft tissue sarcomas can develop in infants
(e.g., infantile fibrosarcoma and malignant hemangiopericytoma). These tumors are
different from the types of soft tissue sarcomas that arise in adolescents (Table IX.2).
♦ Males had slightly higher incidence rates for soft tissue sarcomas than females for
the period 1975-95 (Table IX.3).
♦ Black children had slightly higher incidence rates for soft tissue sarcomas than
white children (Table IX.3), with the largest difference observed among 15-19 year
olds.
♦ The incidence of soft tissue sarcomas among those younger than 20 years of age has
not changed much between 1975-79 (10.2 per million) and 1990-95 (11.3 per million)
(Table IX.4 and Figure IX.5).
Survival
♦ The overall 5-year survival rate for children with rhabdomyosarcoma was approxi-
mately 64% for cases diagnosed from 1985-94 (Figure IX.7). Younger children had
higher survival rates than older children and adolescents, and children with embryo-
nal rhabdomyosarcoma had a more favorable prognosis than children with alveolar
rhabdomyosarcoma (Figure IX.7).
Risk factors
♦ Congenital anomalies and genetic conditions are the only known risk factors
for soft tissue (Table IX.5).

ICCC IX
112
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
INTRODUCTION
The soft tissue sarcomas of childhood
are a heterogeneous group of malignancies
primarily of mesenchymal cell origin that
develop at primary sites throughout the
body [1]. Mesenchymal cells normally
mature into skeletal muscle, smooth
muscle, fat, fibrous tissue, bone and carti-
lage. The malignant counterparts of nor-
mal soft tissue cells include: fibrosarcomas
(fibrous tissue), liposarcomas (adipose
tissue), leiomyosarcomas (smooth muscle),
rhabdomyosarcomas (striated muscle),
angiosarcomas and malignant
hemangiopericytoma (blood vessels), syn-
ovial sarcomas (synovial tissue), and chond-
rosarcomas (cartilage) [1]. Tumors derived
from peripheral nervous system tissues are
also included within the soft tissue sarcoma
category, including malignant peripheral
nerve sheath tumors (also termed malig-
nant schwannoma and
neurofibrosarcoma),and extraosseous
Ewing’s sarcoma [1,2]. The sarcomas of
bone are not included in this discussion, but
are considered within the bone tumor
chapter of this monograph.
In the US, 850-900 children and adoles-
cents younger than 20 years of age are
diagnosed with soft tissue sarcomas each
year, of which approximately 350 are
rhabdomyosarcomas. In children, soft
tissue sarcomas generally are classified as
either rhabdomyosarcomas (RMS) or non-
rhabdomyosarcomas (non-RMS) [1,3,4],
with the non-RMS being further divided
into multiple histologic subtypes such as
those listed in the preceding paragraph [5-
8]. The International Classification of
Childhood Cancer (ICCC) partitions soft
tissue sarcomas into 5 subcategories [9]: a)
the rhabdomyosarcoma subcategory (in-
cluding embryonal and alveolar); b) the
fibrosarcoma subcategory (fibromatous
malignancies and malignant nerve sheath
tumors); c) Kaposi’s sarcoma; d) the “other
specified” soft tissue sarcoma subcategory
(including synovial malignancies; blood
vessel malignancies; myomatous malignan-
cies; lipomatous malignancies; and soft
tissue (extraosseous) Ewing’s sarcoma and
peripheral neuroectodermal tumors) and, e)
the “unspecified” soft tissue sarcoma sub-
category. Individual characteristics of each
subcategory are discussed in more detail in
the sections that follow.
The various soft tissue sarcomas are
associated with distinctive chromosomal
alterations that can be used in some in-
stances to support or confirm a specific
diagnosis [10,11] (Table IX.1). Embryonal
RMS tumor cells often show extra chromo-
some copies (hyperdiploidy) and loss of
heterozygosity involving a specific site on
the short arm of chromosome 11 [11].
Alveolar RMS tumors cells have transloca-
tions involving the FKHR gene on the long
arm of chromosome 13 with genes of the
PAX family on either chromosome 2 (PAX3)
or chromosome 1 (PAX 7) [11]. Many of the
non-RMS also show characteristic chromo-
some translocations. Of note, infantile
fibrosarcoma tumor cells contain the same
chromosomal abnormalities as the tumor
cells of congenital mesoblastic nephroma,
with both possessing t(12;15)(p13;q25)-
associated ETV6-NTRK3 gene fusions [12].
Synovial sarcomas are virtually always
associated with translocations that fuse the
SYT gene on chromosome 18 with the SSX-
1 or SSX-2 genes on the X chromosome [13-
15]. Extraosseous Ewing’s sarcoma and
peripheral neuroectodermal tumors have
translocations involving the EWS gene on
chromosome 22 and either the FLI1 gene
on chromosome 11 or the ERG gene on
chromosome 21 [16]. Malignant peripheral
nerve sheath tumors (also known as
neurofibrosarcomas, malignant
schwannomas, and neurogenic sarcomas)
are associated with neurofibromatosis 1
(NF1) [17], the gene for which is located on
the long arm of chromosome 17 [18]. The
occurrence of characteristic chromosomal
translocations among many of the soft

ICCC IX
113
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
tissue sarcomas of children and adolescents
is in contrast to the rarity of such translo-
cations among the epithelial solid tumors
that predominate among adults, with the
reason(s) for this difference not understood.
INCIDENCE
From 1975-95 in SEER areas, 2,182
neoplasms in children younger than 20
years of age were classified into the ICCC
soft tissue sarcoma diagnostic category.
The ICCC soft tissue sarcoma category is
primarily based on histology and not ana-
tomic site. Thus, nearly one-half of the
cases (974) occurred at anatomic sites other
than connective tissue, with RMS showing
a particular propensity for arising at ana-
tomic sites throughout the body (see RMS
discussion below). Conversely, there were
512 cancers among children arising in
anatomic sites coded as connective tissues
Table IX.1: Molecular characterization of soft tissue sarcomas
Diagnosis Chromosomal Abnormality Genes Involved
Rhabdomyosarcoma, Embryonal
[11]
Hyperdiploidy, and loss-of-
heterozygosity at chromosome
11p15
Unidentified gene at chromosome
band 11p15
Rhabdomyosarcoma, Alveolar
[11]
t(2;13) or t(1;13) FKHR on chromosome 13 and PAX 3
(chromosome 2) or PAX7 (chromosome
1)
Infantile fibrosarcoma
[22,23]
t(12;15) TEL (ETV6) gene on chromosome 12
and NTRK3 (TRKC) on chromosome
15.
Dermatofibrosarcoma protuberans
[24,25]
t(17;22) Platelet-derived growth factor b-chain
(PDGFB) gene on chromosome 17 and
collagen type I alpha 1 (COL1A1) on
chromosome 22
Malignant peripheral nerve sheath
tumors (also known as
neurofibrosarcomas and malignant
schwannomas)
[26,27]
Abnormalities of Chromosome
17
Neurofibromatosis 1 (NF1) gene
Synovial sarcoma
[13-15]
t(X;18) SYT on chromosome 18 and SSX-1 or
SSX-2 on the X chromosome
Liposarcoma
[28-30]
t(12;16), FUS gene on chromosome 16 and
CHOP gene on chromosome 12
Chondrosarcoma, Myxoid
[31,32]
t(9;22) EWS gene on chromosome 22 (also
associated with Ewing’s sarcoma) and
TEC gene on chromosome 9
Extra-osseuous Ewing’s sarcoma
and peripheral neuroectodermal
tumor (PNET) [33]
t(11;22) EWS gene on chromosome 22 and FLI
gene on chromosome 11.
Alveolar soft part sarcoma
[34,35]
t(X; 17) Unidentified gene at chromosome band
17q25
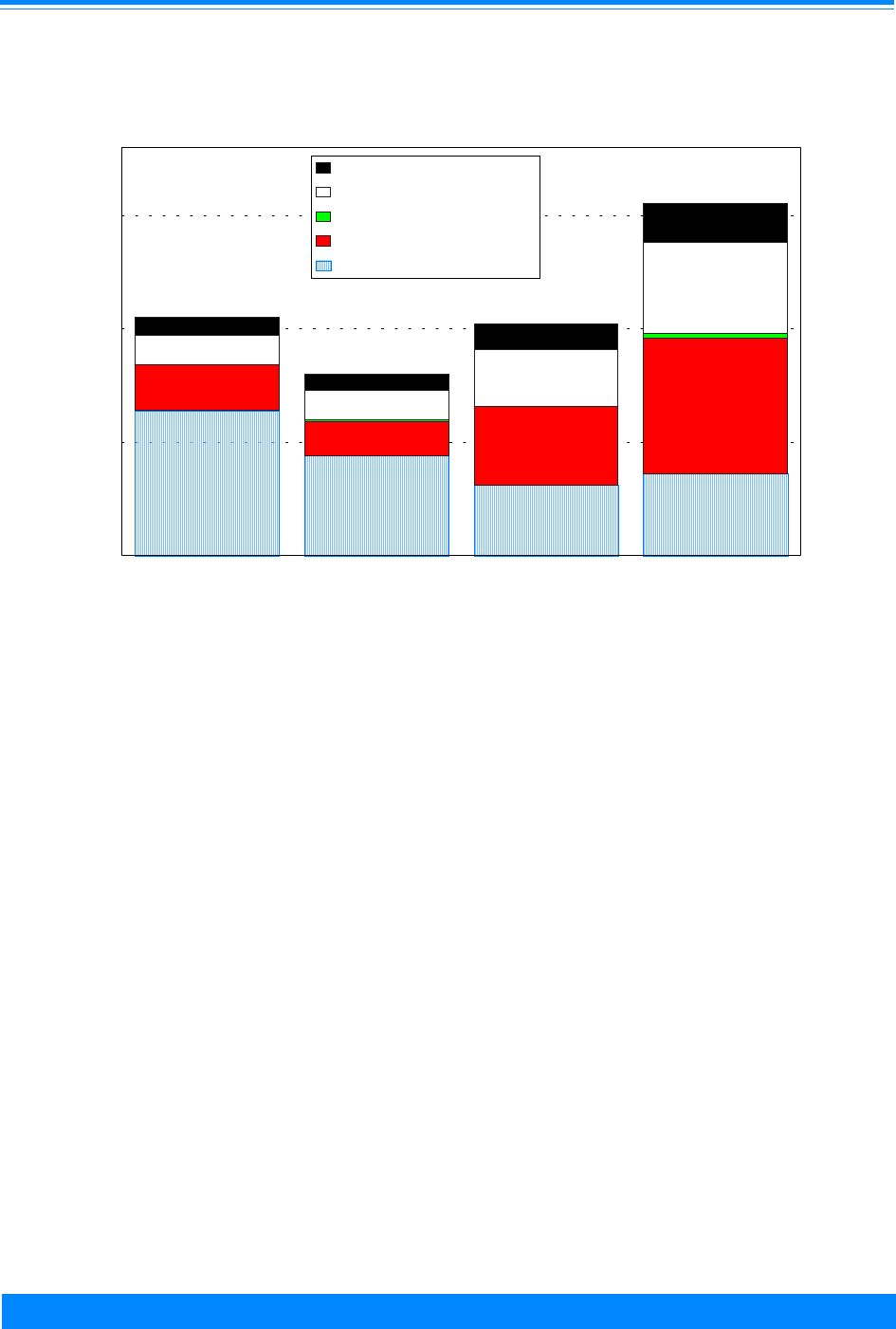
ICCC IX
114
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
that were not included in the ICCC soft
tissue sarcoma category (including 373
classified in the ICCC sympathetic nervous
system tumor category and 77 classified in
the ICCC category germ cell, trophoblastic,
and other gonadal tumor category). These
cases have been included in the appropriate
chapters in the monograph.
Average annual incidence rates of soft
tissue sarcomas are shown in Table IX.2.
Overall, the age-adjusted rate of soft tissue
sarcomas was 11.0 per million children
younger than 20 years of age, which repre-
sented 7% of all primary malignancies for
this population. Of these, 40% were RMS,
29% were in the ICCC fibrosarcoma subcat-
egory, 21% were in the “other specified” soft
tissue sarcoma subcategory, and 10% were
unspecified soft tissue sarcomas. Kaposi’s
sarcoma, a disease associated with AIDS,
was extremely rare in this population, with
only 18 cases reported to SEER areas
during 1975-95.
Histology-specific incidence
Table IX.2 provides the incidence of
specific diagnoses within each of the ICCC
soft tissue sarcoma subcategories. The
incidence of soft tissue sarcoma subtypes
differed notably by age as illustrated in
Figure IX.1. RMS represented 60% of soft
tissue sarcomas for children younger than 5
years of age, but the relative frequency of
RMS decreased with each successive 5-year
age group; RMS accounted for only 23% of
soft tissue sarcomas among the 15-19 year-
old group. The opposite pattern occurred
for the non-RMS subcategories, which
represented 40% of soft tissue sarcomas
among children younger than 5 years of
age, but 77% of these tumors among 15-19
year-olds. The primary diagnoses for each
subcategory are listed and briefly described
below.
The RMS subcategory (ICCC IXa) is
comprised of embryonal and alveolar RMS,
Figure IX.1: Soft tissue sarcoma age-specific incidence rates
by ICCC subcategory, all races, both sexes, SEER, 1975-95
6.4
4.4
3.1
3.6
2
1.5
3.5
6
0.1
0.2
1.3
1.3
2.5
4
0.8
0.7
1.1
1.7
<5 5-9 10-14 15-19
Age (in years) at diagnosis
0
5
10
15
Average annual rate per million
Rhabdomyosarcoma
Fibrosarcoma
Kaposi's sarcoma
Other specified sarcoma
Unspecified sarcoma
10.6
8.0
10.3
15.5

ICCC IX
115
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
Table IX.2: Age-specific and age-adjusted incidence rates per million of soft tissue
sarcomas by ICCC group and subcategory, all races, both sexes, SEER 1975-95
Age (in years) at diagnosis
ICD-O-2 Codes <5 5-9 10-14 15-19 Total*
<15
Total*
<20
Soft Tissue Sarcomas (IX) 10.6 8.0 10.3 15.5 9.6 11.0
Rhabdomyosarcoma
Subcategory (IXa)
6.4 4.4 3.1 3.6 4.6 4.3
Embryonal
rhabdomyosarcoma
8910 4.4 2.7 1.6 1.8 3.0 2.6
Alveolar
rhabdomyosarcoma
8920 0.8 0.8 0.6 0.8 0.7 0.7
Rhabdomyosarcoma,
NOS, pleomorphic, etc.
8900-8902, 8991 1.2 0.9 0.9 0.9 1.0 1.0
Fibrosarcoma
Subcategory (IXb)
2.0 1.5 3.5 6.0 2.3 3.2
Fibrosarcoma 8810 0.3 0.3 0.5 1.1 0.4 0.6
Infantile fibrosarcoma 8814 0.7 0.0 0.0 0.0 0.2 0.2
Malignant fibrous
histiocytoma
8830 0.4 0.4 0.7 1.7 0.5 0.8
Dermatofibrosarcoma 8832 0.2 0.5 1.2 1.9 0.7 1.0
Malignant peripheral
nerve sheath tumor
9540,9560 0.2 0.2 0.8 1.2 0.4 0.6
Kaposi’s sarcoma (IXc) 9140 0 0.1 0 0.2 0 0.1
Other specified STS
Subcategory (IXd)
1.3 1.3 2.5 4.0 1.8 2.3
Liposarcoma 8850,8852,8854 0.1 0.0 0.1 0.4 0.1 0.1
Leiomyosarcoma 8890, 8891 0.1 0.2 0.2 0.7 0.2 0.3
Malignant
mesenchymoma
8990 0.3 0.2 0.1 0.1 0.2 0.2
Synovial sarcoma 9040, 9041 9043 0.1 0.3 0.8 1.4 0.4 0.7
Hemangiosarcoma &
Malignant
Hemangioendothelioma
9120, 9130, 9133 0.1 0.1 0.1 0.3 0.1 0.2
Hemangiopericytoma,
malignant
9150 0.2 0.1 0.1 0.1 0.1 0.1
Alveolar soft part
sarcoma
9581 0.1 0.1 0.1 0.1 0.1 0.1
Chondrosarcoma 9231, 9240 0.0 0.0 0.2 0.0 0.1 0.1
Ewing's (extraosseous)
Family
9364, 9260 0.2 0.3 0.4 0.6 0.3 0.4
Unspecified Subcategory
(IXe)
8800-8804 0.8 0.7 1.1 1.7 0.9 1.1
* Adjusted to the 1970 US standard population
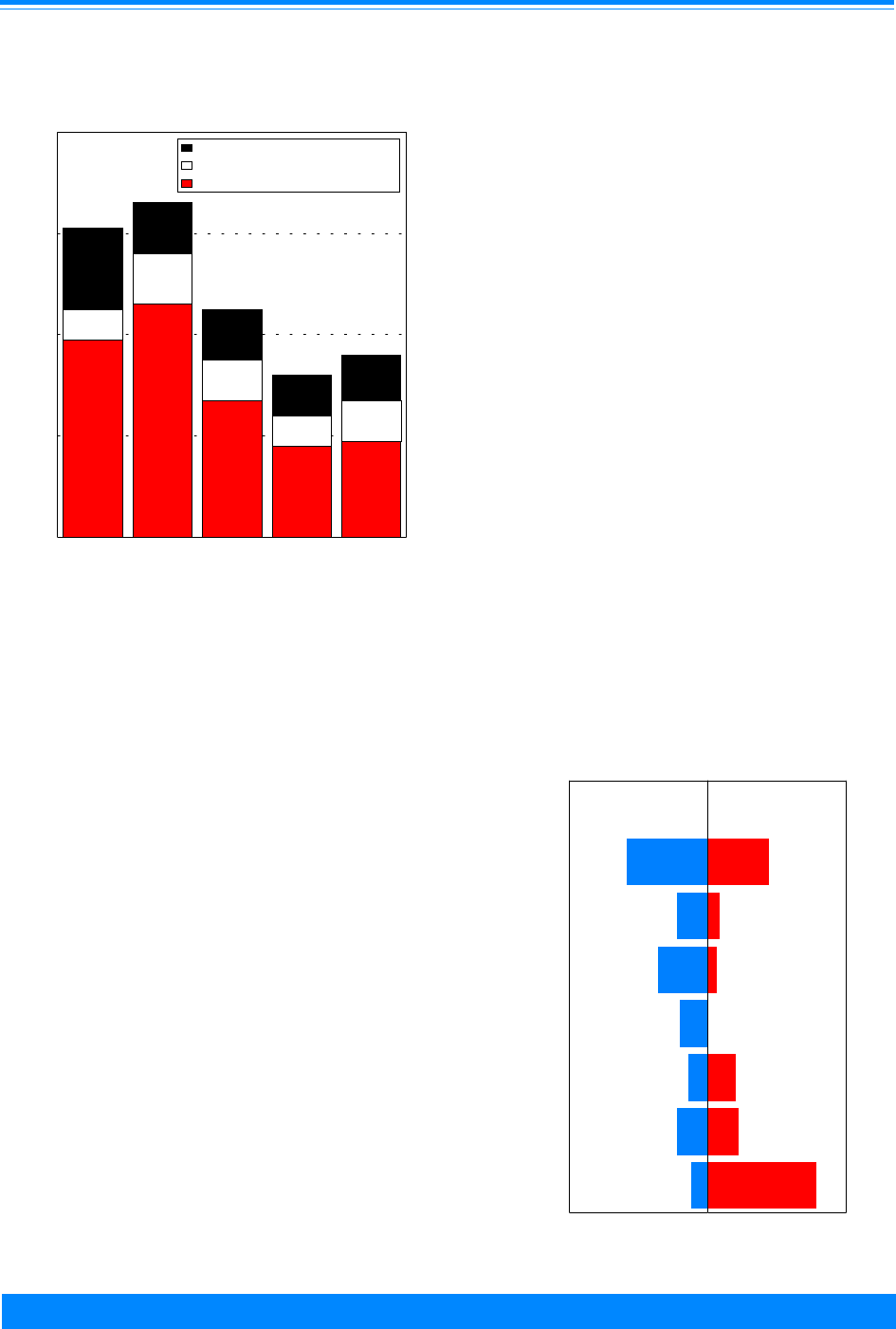
ICCC IX
116
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
as well as “not otherwise specified” RMS,
pleomorphic RMS, mixed-type RMS, and
embryonal sarcoma. RMS ‘not otherwise
specified’ (NOS) represented 17% of all
RMS in SEER areas for 1975-95. Embryo-
nal RMS was the most common type of
RMS at all ages and accounted for 75% of
cases for those younger than 20 years of
age with a specific RMS diagnosis (i.e.,
excluding the NOS category). However, as
shown in Figure IX.2, the incidence of
embryonal RMS varied by age. The rela-
tive percentage of RMS decreased with
increasing age, from 83% of cases with a
specific RMS diagnosis among children
younger than 5 years of age to 64% of cases
among 15-19 year olds. The relative per-
centage of alveolar RMS showed a corre-
sponding increase, from 15% of cases with a
specific RMS diagnosis among children
younger than 5 years of age to 30% of cases
among 15-19 year olds. Pleomorphic (1.5%)
and mixed type RMS (1.0%) comprised only
a small percentage of total RMS.
Embryonal RMS occurred at sites
throughout the body (Figure IX.3), with the
head and neck region (excluding the orbit)
being most common (29% of cases). RMS
arising in the orbit, which is known to have
an especially favorable prognosis [19],
represented an additional 11% of embryo-
nal RMS cases. Genital and urinary organ
sites were also common locations of RMS
development (18% and 10% of embryonal
RMS cases, respectively), while the ex-
tremities were an uncommon site for em-
bryonal RMS (only 6% of embryonal RMS
cases). By comparison, alveolar RMS
occurred most commonly at extremity sites
(39% of alveolar RMS cases) and occurred
infrequently at genitourinary sites (3% of
cases).
The fibrosarcoma subcategory (ICCC
IXb) includes the following diagnoses
(incidence rates for the younger than 20
year old population are provided in paren-
theses): dermatofibrosarcoma (1.0 per
million), malignant fibrous histiocytoma
(0.8 per million), fibrosarcoma (0.6 per
Figure IX.2: Rhabdomyosarcoma (RMS) age-specific
incidence rates by subtype and age group
all races, both sexes, SEER, 1976-84 and 1986-94 combined
3.9
4.6
2.7
1.8
1.9
0.6
1
0.8
0.6
0.8
1.6
1
1
0.8
0.9
< 1 1-4 5-9 10-14 15-19
Age (in years) at diagnosis
0
2
4
6
8
Average annual rate per million
Embryonal
Alveolar
NOS, mixed type, pleomorphic
6.1
6.6
4.4
3.1
3.6
Figure IX.3: Percent distribution of embryonal and
alveolar rhabdomyosarcoma (RMS) by anatomic site
age <20, all races, both sexes, SEER, 1975-95
29
11
18
10
7
11
6
22
4
3
0
10
11
39
Head & Neck
Orbit
Genital
Bladder/
Trunk soft
Pelvic soft
Extremity
Anatomic site
01020304050 0 1020304050
tissue
tissue
(-orbit)
Embryonal
RMS
Alveolar
RMS
Prostate
Relative Percent
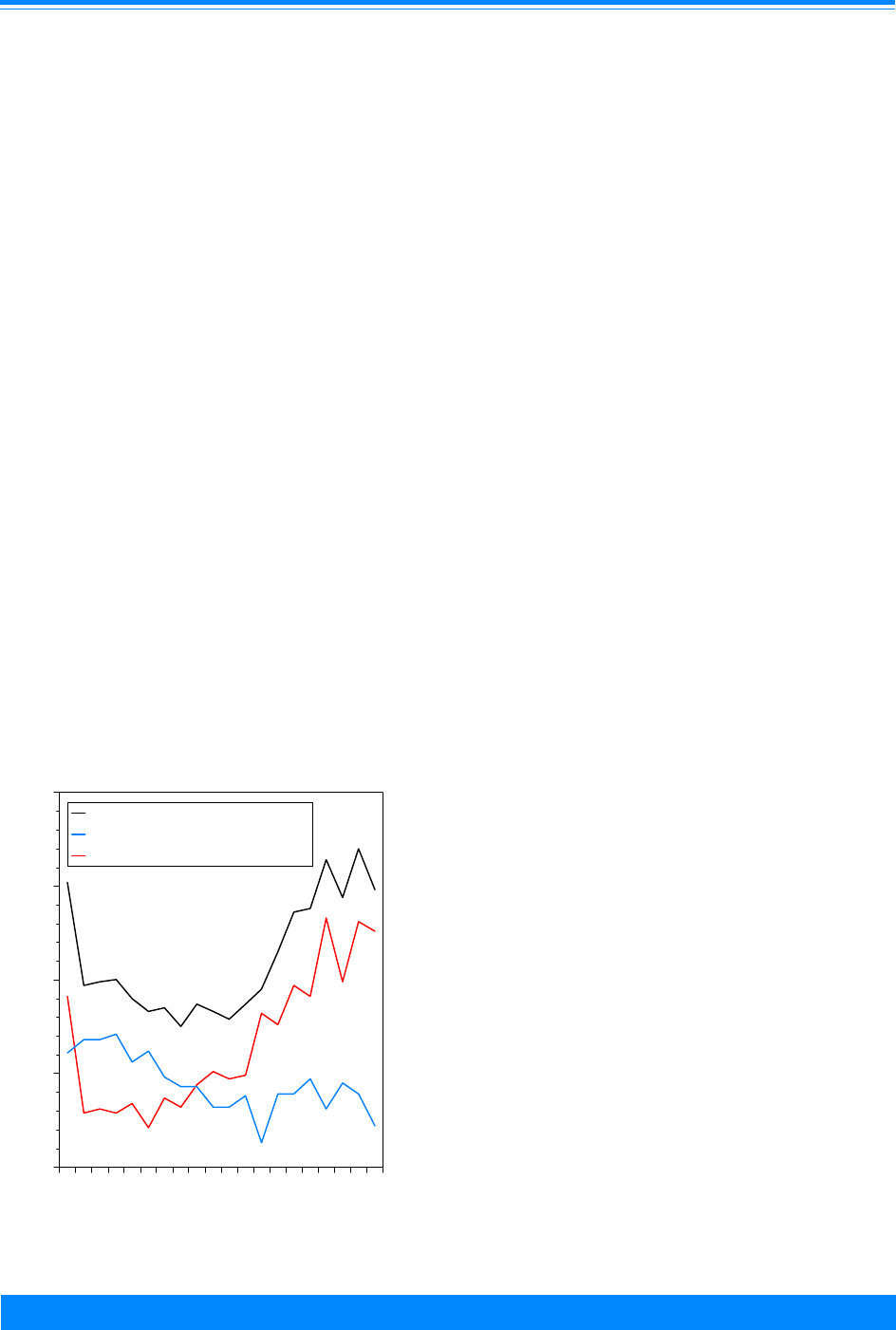
ICCC IX
117
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
million), malignant peripheral nerve sheath
tumor (0.6 per million), and infantile fibro-
sarcoma (0.2 per million). Each of these
soft tissue sarcomas, save infantile fibrosar-
coma, occurs in adults as well as in children
[7,20]. With the exception of infantile
fibrosarcoma, each of these diagnoses
occurred at higher incidence among the 15-
19 year old population than among any of
the younger age groups (Table IX.2). Infan-
tile fibrosarcomas, which are known for
their excellent outcome with surgery alone
[7], occurred only in the younger than 5-
year age group.
For the “other specified” soft tissue
sarcoma subcategory (ICCC IXd), synovial
sarcoma was the most common subtype
(0.7 per million), followed by the Ewing’s
(extraosseous) family of tumors (0.4 per
million) and leiomyosarcoma (0.3 per
million) (Table IX.2). Blood vessel tumors
(e.g., hemangiosarcomas and malignant
hemangiopericytoma), liposarcomas, and
alveolar soft part sarcomas occurred less
commonly. As with the ICCC fibrosarcoma
subcategory, most diagnoses occurred at
higher rates among the 15-19 year old
group than among younger age groups.
Exceptions were malignant
mesenchymoma and malignant
hemangiopericytoma, which developed
most frequently in the first five years of
life.
Age-specific incidence
Figure IX.4 shows incidence rates for
soft tissue sarcomas by single year of age
1
.
Incidence rates were highest among young
children during infancy. Rates dropped in
the second year of life, and remained fairly
stable through age 10 years. After age 10
years, incidence rates began to rise again
as a result of increasing rates for the non-
RMS soft tissue sarcomas. Among infants,
the overall incidence was 15.2 per million,
compared to approximately 10 per million
for children ages 1-4 years. Non-RMS
tumors strongly contributed to the peak in
soft tissue sarcoma incidence during in-
fancy. While RMS accounted for approxi-
mately 40% of soft tissue sarcomas among
infants, RMS occurred at a similar rate
among children 1-4 years. The non-RMS
diagnoses that occurred more commonly in
the first year of life than in the succeeding
4 years included: infantile fibrosarcoma
and fibrosarcoma, NOS (22% of infant soft
tissue sarcomas); malignant
hemangiopericytoma (5% of infant soft
tissue sarcomas), and malignant
mesenchymoma (5% of infant soft tissue
sarcomas).
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.
Figure IX.4: Soft tissue sarcoma age-specific incidence
rates by histology, all races, both sexes
SEER, 1976-84 and 1986-94 combined
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
'
'
''
'
'
'
'
''
''
'
'
''
'
'
'
'
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Age (in years) at diagnosis
0
5
10
15
20
Average annual rate per million
All soft tissue sarcomas
Rhabdomyosarcomas
Non-RMS soft tissue sarcomas
+
'
(

ICCC IX
118
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
Sex-specific incidence
Incidence rates for males and females
are also shown in Table IX.3. Rates among
males tended to be higher than rates for
females within all age groups, although the
overall difference was slight (11.8 per
million males versus 10.3 per million
females for the younger than 20 year old
population). The pattern of rates by age
and histologic subgroups were essentially
the same for males and females.
Black-white differences in incidence
Table IX.3 shows incidence rates by 5-
year age groups for both white and black
children. Black children had slightly higher
incidence rates overall than white children.
Although rate differences were slight
within all age groups, the largest difference
occurred among those 15-19 years of age.
To the extent that numbers allowed reli-
able comparisons, there were no notable
racial differences in soft tissue sarcoma
rates by histologic subgroups.
TRENDS
Average annual age-adjusted incidence
rates across 5-year time periods (6 years for
the last period) are shown in Table IX.4.
Overall rates for soft tissue sarcomas
increased slightly over the first three time
periods from 10.2 to 11.8 per million, and
then dropped a small amount in the
1990-95 period to 11.3 per million. Figure
IX.5 shows the incidence rates for indi-
vidual years from 1975-95 for total soft
tissue sarcomas, RMS, and non-RMS soft
tissue sarcomas. This figure illustrates the
small changes in incidence during this
period; RMS incidence was fairly stable at
4 per million and non-RMS soft tissue
sarcoma incidence varied between 6 and 8
per million.
Table IX.4: Age-adjusted* incidence rates per million of soft tissue sarcomas by time
period, race, and sex, age <20, SEER, 1975-95
1975-79 1980-84 1985-89 1990-95
All races/Both sexes 10.2 10.7 11.8 11.3
Whites
Blacks
10.1
10.2
10.4
10.5
11.5
14.5
10.4
13.9
Males
Females
11.0
9.5
10.6
10.7
13.1
10.5
12.2
10.3
*Adjusted to the 1970 US standard population
Table IX.3: Age-specific and age-adjusted incidence rates per million of soft tissue
sarcomas, by race and sex, SEER, 1975-95
Age (in years) at Diagnosis
ICCC Group <5 5-9 10-14 15-19 <15* <20*
All races/Both sexes
10.6 8.0 10.3 15.5 9.6 11.0
Whites
10.5 7.8 9.9 14.2 9.4 10.6
Blacks 9.6 9.0 11.8 18.9 10.2 12.4
Males 11.2 9.0 10.7 16.2 10.3 11.8
Females
9.9 6.9 9.8 14.7 8.8 10.3
* Adjusted to the 1970 US standard population

ICCC IX
119
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
SURVIVAL
Figure IX.6 shows survival rates for
the time periods 1975-84 and 1985-94. The
5-year relative survival rate for all soft
tissue sarcomas combined was 71% from
1985-1994, with little change from the
earlier period of 1975-84. Survival rates
were higher for the non-RMS fibrosarcoma
subcategory and the “other specified” soft
tissue sarcoma subcategory than for rhab-
domyosarcoma. A small survival improve-
ment in RMS occurred from the earlier to
the later period (59% to 64% 5-year sur-
vival), but no difference between the two
time periods was observed for either the
fibrosarcoma subcategory (82% 5-year
survival) or for the “other specified” soft
tissue sarcoma subcategory (74% 5-year
survival).
Figure IX.6: Soft tissue sarcoma 5-year relative
survival rates, age <20, all races
both sexes, SEER, 1975-84 and 1985-94
69
59
82
74
71
64
82
74
All soft tissue sarcomas (IX)
Rhabdomyosarcomas (IXa)
Fibrosarcoma (IXb)
Other specified sarcomas (IXd)
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Figure IX.5: Trends in total soft tissue sarcoma, rhabdomyosarcoma (RMS)
and non-RMS age-adjusted* incidence rates, age <20, all races, both sexes
SEER 1975-95
*Adjusted to the 1970 US standard population
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
+
+
+
++
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
)
)
)
)
)
)
)
)
)
)
)
))
)
)
)
)
)
)
)
)
1975 1980 1985 1990 1995
Year of diagnosis
0
2
4
6
8
10
12
14
Average annual rate per million
Total STS RMS non-RMS
) + $

ICCC IX
120
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
Additional data on 5-year relative
survival of RMS are shown in Figure IX.7.
Survival among males with RMS was
better than that of females, and survival
was somewhat higher for white children
than for black children. Figure IX.7 also
demonstrates the important prognostic
advantage of younger age. Children
younger than 5 years of age had much
higher 5-year survival rates than 15-19
year olds (79% versus 45%). The prognostic
advantage associated with younger age
may be partially explained by the higher
percentage of embryonal cases among
young children, since RMS cases with
embryonal histology are associated with
superior outcome compared to cases with
alveolar histology (Figure IX.7).
RISK FACTORS
Very little population-based research
has been conducted on potential causes of
RMS or other soft tissue sarcomas in
children. Table IX.5 provides a brief
summary of risk factors that have been
explored. Certain congenital anomalies
and genetic conditions are the strongest
known risk factors, although they explain
only a small proportion of cases. While
the overwhelming majority of RMS occurs
sporadically, a small proportion of RMS is
associated with Li-Fraumeni cancer
susceptibility syndrome (21), and prob-
ably neurofibromatosis type I (3).
SUMMARY
Soft tissue sarcomas accounted for 7%
of all primary malignancies in SEER areas
for children younger than 20 years of age
from 1975-95. RMS represented approxi-
mately 40% of soft tissue sarcomas, with
the remaining non-RMS cases being spread
among multiple diagnoses primarily within
the ICCC fibrosarcoma subcategory and the
“other specific” soft tissue sarcomas subcat-
egory. The average age-adjusted incidence
Figure IX.7: Rhabdomyosarcoma 5-year relative survival rates
by sex, race, subtype, and age, SEER 1985-94
64
69
56
66
57
68
50
79
68
45 45
Total
Male
Female
White
Black
Embryonal
Alveolar
<5
5-9
10-14
15-19
0
20
40
60
80
100
Percent surviving 5 years
Age

ICCC IX
121
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
rate of all soft tissue sarcomas combined
was 11 per million children younger than
20 years of age. While RMS was the most
common soft tissue sarcoma in children,
especially in young children, in older ado-
lescents the non-RMS tumors were more
common than RMS, although no single non-
RMS diagnosis accounted for more than
15% of all cases. There have been very few
population-based studies to evaluate risk
factors for soft tissue sarcoma occurrence;
factors have been identified that explain
only a very small proportion of cases. Five-
year survival rates of soft tissue sarcomas
improved only slightly from the period
1975-84 (69%) to 1985-94 (71%). Children
with RMS had a somewhat poorer 5-year
survival (64%) than did children with non-
RMS in the fibrosarcoma subcategory
(82%) and the “other specified” soft tissue
sarcoma subcategory (74%). Males tended
to have slightly better survival rates than
females, and white children tended to fare
better than black children. Younger chil-
dren with RMS had better outcome than
did older children (Figure IX.7).
Reference List
1. Swanson P, Dehner L: Pathology of soft tissue
sarcomas in children and adolescents. In
Rhabdomyosarcoma and Related Tumors in
Children and Adolescents (HM M, FB R, CE P,
eds). Boca Raton: CRC Press, 1991, pp 385-
419.
2. Brennan M, Casper E, Harrison L: Soft tissue
sarcoma. In Cancer, Principles and Practice of
Oncology (DeVita V, Hellman S, Rosenberg S,
eds). Philadelphia: Lippincott-Raven, 1997, pp
1738-1788.
3. Wexler L, Helman L: Rhabdomyosarcoma and
the undifferentiated sarcomas. In Principles
and Practice of Pediatric Oncology (Pizzo P,
Poplack D, eds). Philadelphia: Lippincott-Raven
Publishers, 1997, pp 799-829.
4. Pappo AS, Shapiro DN, Crist WM, et al: Biology
and therapy of pediatric rhabdomyosarcoma. J
Clin Oncol 13:2123-39, 1995.
5. Pratt C: Clinical manifestations and treatment
of soft tissue sarcomas other than rhabdomyo-
sarcoma. In Rhabdomyosarcoma and Related
Tumors in Children and Adolescents (HM M,
FB R, CE P, eds). Boca Raton: CRC Press,
1991, pp 421-432.
6. Parham DM, Webber BL, Jenkins JJ, 3rd, et al:
Nonrhabdomyosarcomatous soft tissue sarco-
mas of childhood: formulation of a simplified
system for grading. Mod Pathol 8:705-10, 1995.
Table IX.5: Risk factors for soft tissue sarcomas in children
Exposure or Characteristic Comments References
Congenital anomalies There is some concordance with the anatomic location of
RMS and major birth defects. One autopsy study showed
32% of 115 children and adolescents with RMS to have at
least one congenital anomaly.
36,37
Genetic conditions Li-Fraumeni syndrome (associated with p53 mutations),
and neurofibromatosis (associated with NF1 mutations)
21,38,39
Socioeconomic status Low socioeconomic status is associated with increased
risk.
40
Ionizing radiation (in utero) Diagnostic x-rays during pregnancy were associated with
2-fold increase in risk in one study.
41
Parental use of recreational drugs Parents use of marijuana and cocaine during the
pregnancy was associated with increased risk in one
study.
37,42
Known risk factors
Factors for which evidence is
inconsistent or limited

ICCC IX
122
National Cancer Institute
SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
7. Miser J, Triche T, Kinsella T, et al: Other soft
tissue sarcomas of childhood. In Principles and
Practice of Pediatric Oncology (Pizzo P, Poplack
D, eds). Philadelphia: Lippincott-Raven
Publishers, 1997, pp 865-888.
8. Harms D: Soft tissue sarcomas in the Kiel
Pediatric Tumor Registry. Curr Top Pathol
89:31-45, 1995.
9. Kramarova E, Stiller CA: The international
classification of childhood cancer. Int J Cancer
68:759-65, 1996.
10. Hibshoosh H, Lattes R: Immunohistochemical
and molecular genetic approaches to soft tissue
tumor diagnosis: a primer. Semin Oncol 24:515-
25, 1997.
11. Barr FG: Molecular genetics and pathogenesis
of rhabdomyosarcoma. J Pediatr Hematol
Oncol 19:483-91, 1997.
12. Knezevich SR, Garnett MJ, Pysher TJ, et al:
ETV6-NTRK3 gene fusions and trisomy 11
establish a histogenetic link between mesoblas-
tic nephroma and congenital fibrosarcoma.
Cancer Res 58:5046-8, 1998.
13. Kawai A, Woodruff J, Healey JH, et al: SYT-
SSX gene fusion as a determinant of morphol-
ogy and prognosis in synovial sarcoma. N Engl
J Med 338:153-60, 1998.
14. Clark J, Rocques PJ, Crew AJ, et al: Identifica-
tion of novel genes, SYT and SSX, involved in
the t(X;18)(p11.2;q11.2) translocation found in
human synovial sarcoma. Nat Genet 7:502-8,
1994.
15. Crew AJ, Clark J, Fisher C, et al: Fusion of SYT
to two genes, SSX1 and SSX2, encoding
proteins with homology to the Kruppel-
associated box in human synovial sarcoma.
EMBO J 14:2333-40, 1995.
16. Denny CT: Ewing’s sarcoma—a clinical enigma
coming into focus. J Pediatr Hematol Oncol
20:421-5, 1998.
17. Meis JM, Enzinger FM, Martz KL, et al:
Malignant peripheral nerve sheath tumors
(malignant schwannomas) in children [see
comments]. Am J Surg Pathol 16:694-707,
1992.
18. Colman SD, Wallace MR: Neurofibromatosis
type 1. Eur J Cancer 13:1974-81, 1994.
19. Kodet R, Newton WA, Jr., Hamoudi AB, et al:
Orbital rhabdomyosarcomas and related
tumors in childhood: relationship of morphol-
ogy to prognosis—an Intergroup Rhabdomyosa-
rcoma study. Med Pediatr Oncol 29:51-60,
1997.
20. Brennan RJ, Schiestl RH: Chloroform and
carbon tetrachloride induce intrachromosomal
recombination and oxidative free radicals in
Saccharomyces cerevisiae. Mutat Res 397:271-
8, 1998.
21. Malkin D, Li FP, Strong LC, et al: Germ line
p53 mutations in a familial syndrome of breast
cancer, sarcomas, and other neoplasms. Science
250:1233-8, 1990.
22. Knezevich SR, McFadden DE, Tao W, et al: A
novel ETV6-NTRK3 gene fusion in congenital
fibrosarcoma. Nat Genet 18:184-7, 1998.
23. Rubin BP, Chen CJ, Morgan TW, et al: Congeni-
tal mesoblastic nephroma t(12;15) is associated
with ETV6-NTRK3 gene fusion: cytogenetic and
molecular relationship to congenital (infantile)
fibrosarcoma. Am J Pathol 153:1451-8, 1998.
24. Simon MP, Pedeutour F, Sirvent N, et al:
Deregulation of the platelet-derived growth
factor B-chain gene via fusion with collagen
gene COL1A1 in dermatofibrosarcoma
protuberans and giant-cell fibroblastoma. Nat
Genet 15:95-8, 1997.
25. Pedeutour F, Lacour JP, Perrin C, et al: Another
case of t(17;22)(q22;q13) in an infantile der-
matofibrosarcoma protuberans. Cancer Genet
Cytogenet 89:175-6, 1996.
26. Lothe RA, Karhu R, Mandahl N, et al: Gain of
17q24-qter detected by comparative genomic
hybridization in malignant tumors from
patients with von Recklinghausen’s neurofibro-
matosis. Cancer Res 56:4778-81, 1996.
27. Jhanwar SC, Chen Q, Li FP, et al: Cytogenetic
analysis of soft tissue sarcomas. Recurrent
chromosome abnormalities in malignant
peripheral nerve sheath tumors (MPNST).
Cancer Genet Cytogenet 78:138-44, 1994.
28. Willeke F, Ridder R, Mechtersheimer G, et al:
Analysis of FUS-CHOP fusion transcripts in
different types of soft tissue liposarcoma and
their diagnostic implications. Clin Cancer Res
4:1779-84, 1998.
29. Rabbitts TH, Forster A, Larson R, et al: Fusion
of the dominant negative transcription regula-
tor CHOP with a novel gene FUS by transloca-
tion t(12;16) in malignant liposarcoma. Nat
Genet 4:175-80, 1993.
30. Crozat A, Aman P, Mandahl N, et al: Fusion of
CHOP to a novel RNA-binding protein in
human myxoid liposarcoma. Nature 363:640-4,
1993.
31. Labelle Y, Zucman J, Stenman G, et al: Onco-
genic conversion of a novel orphan nuclear
receptor by chromosome translocation. Hum
Mol Genet 4:2219-26, 1995.
32. Clark J, Benjamin H, Gill S, et al: Fusion of the
EWS gene to CHN, a member of the steroid/
thyroid receptor gene superfamily, in a human
myxoid chondrosarcoma. Oncogene 12:229-35,
1996.
33. Denny CT: Gene rearrangements in Ewing’s
sarcoma. (47 Refs). Cancer Invest 14:83-8,
1996.
34. Heimann P, Devalck C, Debusscher C, et al:
Alveolar soft-part sarcoma: further evidence by
FISH for the involvement of chromosome band
17q25 [In Process Citation]. Genes Chromo-

ICCC IX
123
National Cancer Institute SEER Pediatric Monograph
SOFT TISSUE SARCOMAS
somes Cancer 23:194-7, 1998.
35. Sciot R, Dal Cin P, De Vos R, et al: Alveolar
soft-part sarcoma: evidence for its myogenic
origin and for the involvement of 17q25.
Histopathology 23:439-44, 1993.
36. Yang P, Grufferman S, Khoury MJ, et al:
Association of childhood rhabdomyosarcoma
with neurofibromatosis type I and birth
defects. Genetic Epidemiol 12:467-74, 1995.
37. Ruymann F, Grufferman S: Introduction and
epidemiology of soft tissue sarcomas. In
Rhabdomyosarcoma and Related Tumors in
Children and Adolescents (HM M, FB R, CE P,
eds). Boca Raton: CRC Press, 1991, pp 3-18.
38. Li FP, Fraumeni JF, Jr.: Soft tissue sarcomas,
breast cancer, and other neoplasms. A familial
syndrome? Ann Intern Med 71:747-52, 1969.
39. Hartley AL, Birch JM, Marsden HB, et al:
Neurofibromatosis in children with soft tissue
sarcoma. Pediatr Hematol Oncol 5:7-16, 1988.
40. Grufferman S, Wang HH, DeLong ER, et al:
Environmental factors in the etiology of
rhabdomyosarcoma in childhood. J Natl Cancer
Inst 68:107-13, 1982.
41. Grufferman S, Gula M, Olshan A, et al: In
utero X-ray exposure and risk of childhood
rhabdomyosarcoma. Paediatr Perinat
Epidemiol 5:A6, 1991.
42. Grufferman S, Schwartz AG, Ruymann FB, et
al: Parents’ use of cocaine and marijuana and
increased risk of rhabdomyosarcoma in their
children. Cancer Causes Control 4:217-24,
1993.

124
National Cancer Institute
SEER Pediatric Monograph

ICCC X
125
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
HIGHLIGHTS
Incidence
♦ While germ cell, trophoblastic and other gonadal (GCTOG) tumors represented 16%
of all cancers among adolescents between 15 and 19, they represented only 7% of
cancer diagnoses among children younger than 20 (incidence 12.0 per million) and
3.5% of cancer diagnoses for children younger than 15 (incidence 5.4 per million)
(Table X.4).
♦ In the US, approximately 900 children and adolescents younger than 20 years of age
are diagnosed with germ cell tumors each year.
♦ The majority (61%) of GCTOG tumors occurring among children younger than 20
years are gonadal (ovarian or testicular) germ cell tumors (Table X.1). However,
when only children younger than 15 years of age are considered, non-gonadal germ
cell tumors are more common than gonadal germ cell tumors (Table X.4).
♦ For males, the incidence rates of testicular (Xc) and non-CNS extragonadal (Xb)
germ cell tumors were similar during the first year of life at approximately 9 per
million, and then declined to very low levels by age 4. Between ages 4 and 15 the
rates of testicular germ cell tumors remained very low, but between ages 15 and 19
years of age, the incidence rates increased dramatically (Figure X.2).
♦ For females, ovarian (gonadal) germ cell tumors (Xc) began to increase in incidence
at age 8-9 years and peaked at age 18 (20 per million) (Figure X.3). For males, the
rate of testicular germ cell tumors (Xc) at age 19 was substantially higher than that
observed for ovarian germ cell tumors among 19 year old females (44.5 versus 10.4
per million).
♦ White males younger than age 20 had much higher rates of testicular germ cell
tumors (9.1 per million) than blacks males (1.2 per million). In contrast, white
females younger than age 20 had slightly lower rates (4.5 per million) than black
females (5.6 per million) for ovarian germ cell tumors (Table X.3).
Survival
♦ For patients younger than 20 years of age, females had slightly higher 5-year
survival rates than males, and whites had somewhat higher 5-year survival rates
than blacks for GCTOG tumors (Figure X.6).
♦ Increasing survival rates were observed between 1975-84 and 1985-94 for each
subgroup of the ICCC for patients younger than 20 (Figure X.7). The overall 5-year
relative survival rate for all subgroups combined increased from 77% to 87% (Figure
X.6).
♦ The increase in survival between 1975-84 and 1985-94 was similar for ovarian and
testicular germ cell tumors. Both increased from 82% to 93-94% (Figure X.7). Young
males (<5 years) survived better than males aged 15-19.
Risk factors
♦ The etiology of malignant germ cell tumors is poorly understood. Cryptorchidism is
the only confirmed risk factor for testicular germ cell tumors (see Table X.5 for
references).
Leslie Bernstein, Malcolm A. Smith, Lihua Liu, Dennis Deapen, Debra L. Friedman
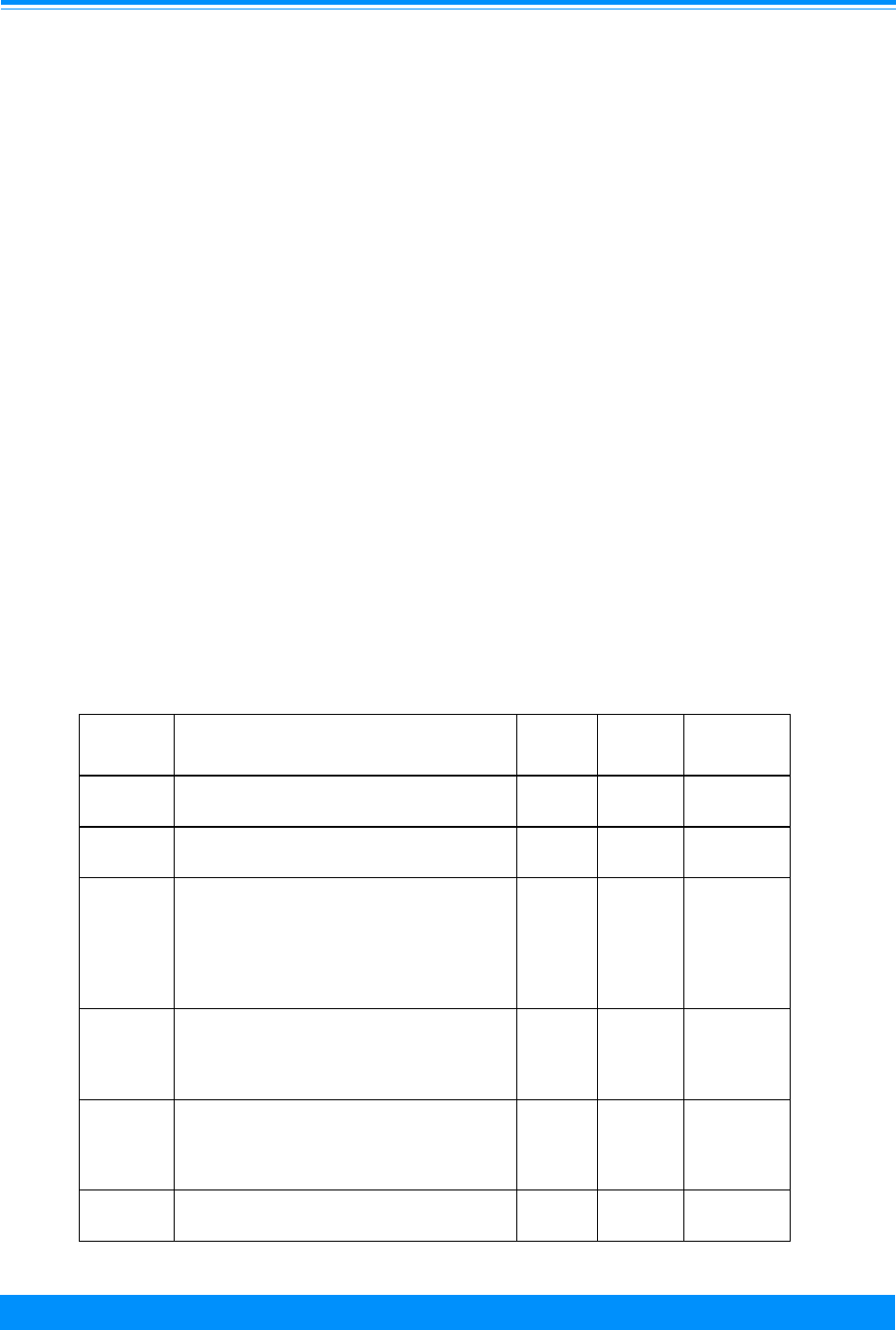
ICCC X
126
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
INTRODUCTION
Germ cell tumors are biologically
diverse and histologically heterogeneous [1-
3], with a substantial proportion having
benign rather than malignant behavior
(particularly among young children). Germ
cell tumors originate in primordial germ
cells, which may undergo germinomatous or
embryonic differentiation. Primordial germ
cells are initially detectable in the yolk sac
of the four week embryo, and their migra-
tory route during embryogenesis from the
yolk sac to the gonads (either the testes or
ovaries) may account for the primarily mid-
line location of most extragonadal germ cell
tumors [1].
Germ cell tumors are grouped together
with trophoblastic and other gonadal
neoplasms in the International Classifica-
tion of Childhood Cancer (ICCC) [4]. For
shorthand notation this entire group, ICCC
X, will be abbreviated as GCTOG tumors.
This diagnostic group is categorized into
five subgroups according to the cells of
origin of the cancer (germ cells, trophoblas-
tic cells or other cells) and the location in
the body of the cancer (gonads: testes or
ovaries; central nervous system; or else-
where) (see Table X.1).
In the US, approximately 900 children
and adolescents younger than 20 years of
age are diagnosed with germ cell tumors
each year. Essential for understanding the
incidence patterns for germ cell tumors of
children and adolescents is recognition that
the germ cell tumors of infancy and early
childhood are biologically distinctive from
those that arise in older children and
adolescents [2,3]. Thus, tumors in the same
ICCC subgroup may have very different
biological characteristics and clinical be-
havior (Table X.2. [5]). The categorization
of germ cell tumors in Table X.2 provides a
Table X.1: Average annual age-adjusted* incidence rates per million for germ cell
trophoblastic and other gonadal cancers by sex and subtype, age <20
all races, SEER, 1986-95
ICCC
Group X
Description Total Males Females
X a-e Germ cell, trophoblastic and other
gonadal tumors
11.6 12.0 11.1
Xa Intracranial and intraspinal germ cell
tumors
1.6 2.3 0.9
Xb Other and unspecified non-gonadal germ
cell tumors. (This category includes the
tumors of infants and young children that
originate in the sacrococcygeal region, as
well as mediastinal tumors primarily
developing in older children.)
1.6 1.5 1.8
Xc Gonadal germ cell tumors
Testis
Ovary
6.7
4.1
2.6
8.0
8.0
-
5.3
-
5.3
Xd Gonadal carcinoma
Ovary
Other
1.4
1.3
0.1
0.1
-
0.1
2.9
2.6
0.3
Xe Other and unspecified malignant gonadal
tumors
0.2 0.1 0.3
*Adjusted to the 1970 US standard population

ICCC X
127
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
Table X.2: GCTOG tumors by sub-group, age and biological characteristics [5]
GCTOG
TUMORS
(ICCC X)
Site Age Characteristics
Intracranial and
intraspinal germ
cell tumors
(ICCC Xa)
Intracranial
(especially pineal
region) [2]
Older children,
adolescents and
adults
Some, though not all, of these tumors
have biological characteristics similar to
those of testicular germ cell tumors in
adolescents and young adults (e.g., an
isochromosome of the short of
chromosome 12 as discussed below) [6-9].
Non-CNS, Non-
gonadal germ cell
(ICCC Xb)
Sacrococcygeal/pel
vic region [2]
Infants and young
children
The biological characteristics of these
tumors is similar to those of testicular
germ cell tumors in young boys (see
below), but different from those of
testicular germ cell tumors in adolescents
and young adults (see below).
“ Mediastinum [2] Older children,
adolescents and
adults
Some, though not all, mediastinal germ
cell tumors have biological characteristics
similar to those of testicular germ cell
tumors in adolescents and young adults
(e.g., an isochromosome of the short of
chromosome 12 as discussed below)
[10,11].
Gonadal germ cell
(ICCC Xc)
Testicular Infants and young
boys
The biological characteristics of these
tumors are distinctive from those of
testicular germ cell tumors in adolescents
and young adults (see below). The
tumors primarily show yolk sac tumor
(endodermal sinus tumor) histology and
are generally diploid or tetraploid.
Recurring chromosomal abnormalities
include deletions of chromosome 1p and
6q, but not isochromosome of the short
arm of chromosome 12 [12-15].
“ Testicular Adolescents and
young adults
These typically possess an
isochromosome of the short arm of
chromosome 12 [5,16-19] and are
aneuploid [12,19]
“ Ovary Adolescents and
adults
These show greater biological diversity
than do germ cell tumors arising in the
testes, and include malignant teratomas
and other malignant germ cell tumors
(e.g., dysgerminomas, yolk sac tumors,
and mixed germ cell tumors). Like their
testicular counterparts, they commonly
show increased copies of the short arm of
chromosome 12 [5].
Gonadal
carcinomas
(Xd)
Ovary Adolescents and
adults
These carcinoma tumors are not
biologically related to the germ cell
tumors and develop almost exclusively in
the ovary.

ICCC X
128
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
basis for understanding the incidence
patterns and trends of germ cell tumors in
children.
A total of 2,065 children younger than
20 years of age were diagnosed with
GCTOG tumors during the period 1975
through 1995 in the SEER areas. This
represents 7% of all neoplasms diagnosed
among children younger than 20 years of
age: 3.5% of all neoplasms for children
younger than 15 years of age and a much
higher proportion, 13.9%, for 15-19 year
olds. The majority (1,260 or 61%) of
GCTOG tumors occurring among children
younger than 20 years of age are gonadal
(ovarian or testicular) germ cell tumors
(Xc). However, when only children younger
than 15 years of age are considered, non-
gonadal germ cell tumors (Xa and Xb) are
more common than gonadal germ cell
tumors.
The GCTOG tumor group (ICCC X)
includes 94% of the malignant testicular
tumors and 99% of the ovarian tumors
among children and adolescents. Six
percent of malignant testicular tumors and
less than 1% of ovarian tumors are sarco-
mas and are grouped under ICCC IX (soft
tissue sarcomas). Excluding the sarcomas,
nearly all of the testicular tumors in male
children and adolescents were germ cell
tumors, 98%. Excluding the small number
of ovarian sarcomas, the histologic types of
ovarian tumors in female children and
adolescents were 64% germ cell (Xc), 33%
carcinomas (Xd), and 3% other and unspeci-
fied (Xe).
INCIDENCE
Sex-specific incidence
Table X.1 shows the incidence of
GCTOG tumors by sex for children younger
than 20 years of age for the years 1986 to
1995. The incidence for males (12.0 per
million) slightly exceeded that for females
(11.1 per million). For males, the subgroup
with the highest incidence was testicular
germ cell tumors (8.0 per million). For
females, ovarian germ cell tumors had the
highest rate (5.3 per million). Intracranial
and intraspinal germ cell tumors (ICCC Xa)
were more common in males (2.3 per mil-
lion) than in females (0.9 per million), and
accounted for about 14 percent of all
GCTOG tumors among those younger than
20 years of age. Non-gonadal germ cell
tumors arising outside of the central ner-
vous system (CNS), ICCC Xb, occurred with
similar frequency among males and fe-
males. In contrast gonadal carcinomas
were almost exclusively seen among fe-
males and most of these were ovarian
gonadal carcinomas.
Table X.3: Average annual age-adjusted* incidence rates per million for germ cell
trophoblastic and other gonadal cancers by race, sex, and subtype
age <20, SEER, 1975-95
ICCC
Group X
ICCC Germ Cell Tumor
Category
White
Male
Black
Male
White
Female
Black
Female
X a-e All 12.3 3.2 9.0 10.8
Xc Gonadal germ cell tumors 9.1 1.2 4.5 5.6
Testis 9.1 1.2 - -
Ovary - - 4.5 5.6
X a,b,d,e Other than gonadal germ cell
tumors
3.2 2.0 4.5 5.2
*Adjusted to the 1970 US standard population

ICCC X
129
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
Black-white differences in incidence
Black children had a lower incidence of
germ cell tumors than white children (7.0
vs. 10.7 per million). This difference was
primarily the result of a lower rate of
gonadal germ cell tumors among blacks
than whites. Table X.3 shows the incidence
rates for gonadal germ cell tumors for
children younger than 20 years of age for
the years 1975 to 1995 by race and sex.
Remarkably, the lower rates of gonadal
germ cell tumors among black children
were restricted to males. For children
younger than 20 years of age, black males
had a rate of testicular germ cell tumor
that was only one-seventh that for white
males (1.2 versus 9.1 per million), while
black females had slightly higher rates of
ovarian germ cell tumors than white fe-
males (5.6 versus 4.5 per million). The low
rate of testicular germ cell tumors observed
among young black males is consistent
with the reported low incidence for testicu-
lar cancer among adult black males [20-22].
Age-specific incidence
Figure X.1 shows the age-specific inci-
dence of GCTOG tumors by single year of
age and sex.
1
Rates were relatively high in
the first year of life and then declined to
very low levels before increasing at age 8-
12 years for females and at age 11-14 years
for males. Incidence continued to increase
for both males and females up through age
19. The distribution of tumor types by age
was distinctive for males and females.
For males, the incidence rates of testicu-
lar (Xc) and non-CNS extragonadal (Xb)
germ cell tumors were similar during the
first year of life at approximately 9 per
Figure X.1: GCTOG age-specific incidence rates
by sex, all races, SEER, 1986-94
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
50
Average annual rate per million
Males
Females
"
)
GCTOG - Germ cell, trophoblastic and other gonadal
1
Enumeration of the population at risk by single years of age was
available only for the census year 1990. The US Bureau of the
Census provides intercensal population estimates by 5-year age
groups, but not by single years of age. Therefore, the population
estimates for 1990 were used in rate calculations for cases
diagnosed from 1986-94.

ICCC X
130
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
million, and then declined to very low levels
by age 4 years (Figure X.2). Between ages
4 and 15 the rates remained very low, but
between ages 15 and 19 years of age, the
incidence rates increased dramatically.
For females, non-CNS extragonadal
germ cell tumors (Xb) accounted for the
vast majority of cases in the first year of
life, with ovarian germ cell tumors (Xc)
being extremely rare (Figure X.3). Most of
the extragonadal germ cell tumors arising
in the first year of life occurred in pelvic
soft tissue (e.g., the sacrococcygeal region)
and in the retroperitoneum. Gonadal germ
cell tumors (Xc) began to increase in inci-
dence for females at age 8-9 years, while
gonadal carcinomas (Xd) began to increase
after age 12. By age 19, the rate of gonadal
carcinomas (Xd) was similar to ovarian
germ cell tumors (Xc) in females. For
males, the rate of testicular germ cell
tumors (Xc) at age 19 was substantially
higher than that observed for ovarian germ
cell tumors among 19 year old females
(44.5 versus 10.4 per million for age 19).
TRENDS
The age-adjusted incidence rates for
GCTOG tumors increased between 1975-79
and 1990-95 from 3.7 to 5.4 per million for
children younger than 15 years of age and
from 8.5 to 12.0 per million for those
younger than 20 years of age (Table X.4).
For both males and females younger than
15 years of age, the increase in incidence
primarily resulted from higher rates for
intracranial and intraspinal germ cell
tumors (Xa) and for non-CNS extragonadal
germ cell tumors (Xb), while the rates of
gonadal germ cell tumors (Xc) did not
increase. The increased incidence of non-
CNS extragonadal tumors (Xb) for both
males and females was due in large mea-
sure to an increase in incidence in the first
Figure X.2: GCTOG age-specific incidence rates
by selected ICCC subgroups, males
all races, SEER, 1986-94
"
"
"
"
"
"
"
"
""""""
"
"
"
"
"
"
'
'
'
'
''
'
'''''
'
'
'
'
'
'
'
'
+
+
+
+++
+
+
+
+
+
+
+
+
+
+
+
+
+
+
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
Average annual rate per million
Xa = Intracranial/intraspinal germ cell tumors
Xb = Other non-gonadal germ cell tumors
Xc = Testicular germ cell tumors
+
'
"
GCTOG - Germ cell, trophoblastic and other gonadal
Figure X.3: GCTOG age-specific incidence rates
by selected ICCC subgroups
females, all races, SEER, 1986-94
,
,
,,,,,,,,,
,,
,
,
,
,
,
,
,
"
"
"
""
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
'
'
'
'
'
''
''
'
'
'''
'
'
''
'
'
+
+
+
+++
+
+
+
+
+
+
+
+
+
+
+
+
+
+
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
Average annual rate per million
Xa = Intracranial/intraspinal germ cell tumors
Xb = Other non-gonadal germ cell tumors
Xc = Ovarian germ cell tumors
Xd = Gonadal carcinoma
+
'
"
,
GCTOG - Germ cell, trophoblastic and other gonadal
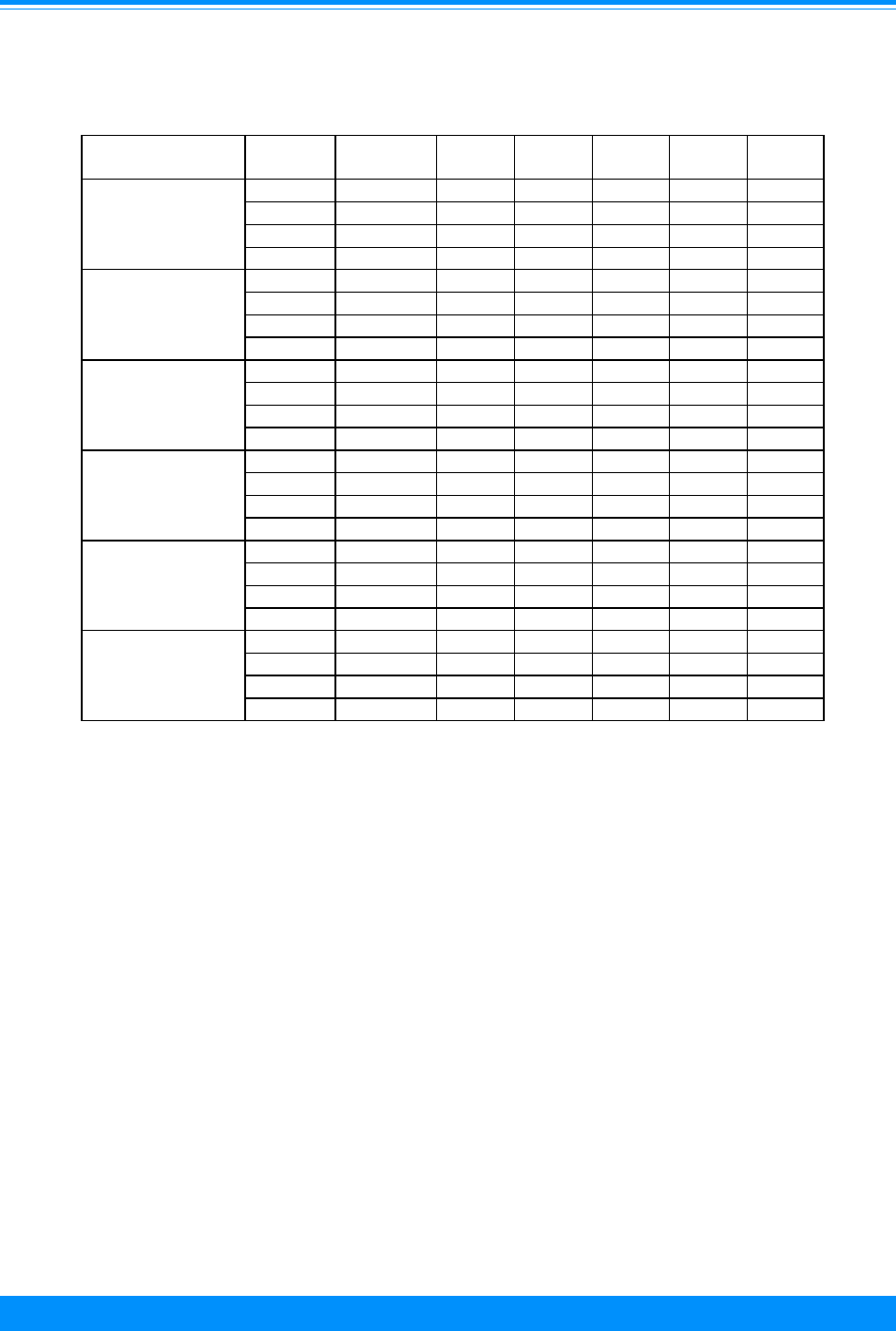
ICCC X
131
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
year of life. This increase in non-CNS
extragonadal malignant tumors among
infants must be interpreted with caution,
because non-malignant sacrococcygeal
teratomas diagnosed in the newborn period
outnumber malignant teratomas [3,23-25],
and because careful inspection of mature
and immature sacrococcygeal teratomas
may show microscopic foci of yolk sac tumor
[26,27]. Since nonmalignant sacrococcygeal
teratomas are not reported and yolk sac
tumors are reported, the increase in inci-
dence in the first year of life may be the
result of increasing recognition by patholo-
gists of the need for careful scrutiny of
apparently non-malignant sacrococcygeal
teratomas. Almost all of the increase in the
first year of life for females was in malig-
nant teratomas/embryonal teratomas.
An increase in the age-adjusted inci-
dence for GCTOG tumors was also observed
for both sexes among those younger than 20
years of age. For males younger than 20
years of age, the increase in incidence was
from 9.1 to 12.2 per million, with most of
the increase attributed to intracranial and
intraspinal germ cell tumors (Category Xa)
and to testicular germ cell tumors (Category
Xc). For females, the increase was from 7.8
to 11.7 per million, with most of the increase
attributable to ovarian germ cell tumors
Table X.4:Average annual age-adjusted
1
incidence rates per million for germ cell
trophoblastic, and other gonadal cancers by sex, age, subtype, and
time period, all races, SEER, 1975-95
Sex/Age Group Years X(total) Xa
2
Xb
2
Xc
2
Xd
2
Xe
2
Total <15 1975-79 3.7 0.5 0.7 2.2 0.1 0.2
1980-84 4.8 0.9 1.1 2.6 0.1 0.1
1985-89 4.8 0.7 1.3 2.7 0.1 0.1
1990-95 5.4 1.5 1.4 2.1 0.3 0.0
Males <15
1975-79 3.1 0.5 0.6 1.8 0.1 0.1
1980-84 4.1 1.2 0.9 2.0 0.0 0.0
1985-89 4.1 1.1 0.8 2.2 0.0 0.1
1990-95 4.4 1.9 1.1 1.4 0.0 0.0
Females <15
1975-79 4.3 0.4 0.8 2.6 0.2 0.4
1980-84 5.5 0.6 1.3 3.3 0.2 0.2
1985-89 5.6 0.4 1.8 3.2 0.1 0.1
1990-95 6.4 1.2 1.9 2.7 0.7 0.0
Total <20
1975-79 8.5 0.6 1.4 5.4 0.8 0.3
1980-84 9.6 0.9 1.5 6.4 0.7 0.2
1985-89 10.7 1.1 1.7 6.6 1.1 0.2
1990-95 12.0 1.9 1.6 6.8 1.6 0.2
Males <20
1975-79 9.1 1.0 1.2 6.9 0.1 0.1
1980-84 11.0 1.2 1.5 8.1 0.1 0.1
1985-89 11.4 1.7 1.6 7.8 0.1 0.1
1990-95 12.2 2.6 1.3 8.1 0.1 0.1
Female <20 1975-79 7.8 0.3 1.6 3.9 1.5 0.6
1980-84 8.1 0.6 1.5 4.5 1.3 0.3
1985-89 10.0 0.4 1.8 5.3 2.2 0.3
1990-95 11.7 1.1 1.8 5.3 3.2 0.3
1
Adjusted to the 1970 US standard population
2
Xa = Intracranial and intraspinal germ cell tumors; Xb = Other and unspecified
non-gonadal germ cell tumors; Xc = Gonadal (ovarian and testicular) germ cell tumors;
Xd = Gonadal carcinoma; Xe = Other and unspecified malignant gonadal tumors.

ICCC X
132
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
(Category Xc) and to ovarian carcinomas
(Category Xd). Because of the larger num-
ber of cases in the 15-19 year group com-
pared to the younger than 15 year group,
the trends for those younger than 20 years
of age are primarily determined by trends
for the 15-19 year age group.
Figure X.4 illustrates the increase in
incidence of testicular germ cell tumors (Xc)
for the 15-19 year age group between 1975-
79 (22 per million) and 1990-1995 (28 per
million). The increase in incidence of
testicular germ cell tumors for those 15-19
years of age is reminiscent of the increase
in testicular cancer among adult males.
Over the past 30-40 years, increased rates
of testicular cancer have been reported
from developed countries throughout the
world, including the United States [21,22],
European countries [28], Australia [29],
and New Zealand [30].
The overall rate of GCTOG tumors for
females aged 15-19 increased markedly
from 1975-79 to 1990-95 (Figure X.5), but
much of the increase was attributable to
the inclusion of borderline tumors of the
ovary which were not reportable cancers for
the entire time period. Figure X.5 shows
the overall rate for ICCC X for females both
with and without the borderline tumors.
With the borderline tumors excluded, the
overall rate increased only slightly between
1975-79 and 1990-95 (Figure X.5), and this
increase was driven by an increased inci-
dence of ovarian germ cell tumors (8 per
million for 1975-79 to 13 per million for
1990-95). An increased incidence for ova-
rian germ cell tumors in adults has also
been reported [31,32].
SURVIVAL
For the period from 1985 to 1994,
the 5-year survival rate for patients
Figure X.4: Trends in GCTOG age-specific
incidence rates by selected ICCC subgroups
age 15-19, males, all races, SEER 1975-95
-
-
-
-
%
%
%
%
$
$
$
$
#
#
#
#
1975-79 1980-84 1985-89 1990-95
Year of diagnosis
0
10
20
30
40
Average annual rate per million
Total X
Xa = Intracranial and intraspinal germ cell tumors
Xb = Other and unspecified non-gonadal germ cell tumors
Xc = Gonadal (testicular) germ cell tumors
#
$
%
-
GCTOG - Germ cell, trophoblastic and other gonadal
Figure X.5: Trends in GCTOG age-specific
incidence rates by selected ICCC subgroups
age 15-19, females, all races, SEER, 1975-95
'
'
'
'
'
'
'
'
)
)
)
)
-
-
-
-
$
$
$
$
#
#
#
#
1975-79 1980-84 1985-89 1990-95
Year of diagnosis
0
10
20
30
40
Average annual rate per million
Total X
excluding borderline
Xb = Other & unspecified non-gonadal germ cell tumors
Xc = Gonadal (ovarian) germ cell tumors
Xd - borderline
Xd - other
#
$
-
)
'
'
GCTOG - Germ cell, trophoblastic and other gonadal

ICCC X
133
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
younger than 20 years of age with germ cell
tumors was 87% (Figure X.6). Survival
rates were better for the 15-19 year olds (5-
year survival, 90%) than for the younger
than 15 year olds (5-year survival, 84%).
Other observations about outcome for
children with germ cell tumors are
illustrated in Figures X.6 and X.7 and
include:
• For those younger than 20 years of
age, females had slightly higher 5-
year survival rates than males, and
whites had somewhat higher 5-year
survival rates than blacks (Figure
X.6).
• Survival for patients younger than
20 years of age was better for
gonadal germ cell tumors (ICCC Xc)
than for tumors arising at “other
and unspecified” sites (ICCC
Category Xb), with 5–year survival
rates of 94% and 71%, respectively
in 1985-1994. Outcome was similar
for patients younger than 20 years
of age with intracranial germ cell
tumors (ICCC Xa) and with tumors
arising at “other and unspecified”
sites (ICCC Xb), with both groups
having survival rates for 1985-94 of
approximately 70% (Figure X.7).
• Increasing survival rates were
observed between 1975-84 and
1985-94 for each subgroup of the
ICCC for patients younger than 20
years if age. The overall 5-year
relative survival rate for all
subgroups combined increased from
77% to 87% (Figure X.6). The
largest increase in survival was for
tumors arising at other and
unspecified sites (ICCC Xb): 58
percent compared to 72 (Figure X.7).
Figure X.6: Germ-cell tumor 5-year relative survival
rates by sex, race, age, and time period
SEER (9 areas), 1975-84 and 1985-94
GCTOG - Germ cell, trophoblastic and other gonadal
77
74
81
77
82
61
71
82
79
88
86
89
88
84
82 82 82
90
Total Male Female White Black <5 5-9 10-1415-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race Age
Figure X.7: GCTOG tumor 5-year relative survival rates
by sub-group, sex and time period, all races
SEER (9 areas), 1975-84 and 1985-94
# - < 25 cases - rate not shown
GCTOG - Germ cell, trophoblastic and other gonadal
Xa = Intracranial and intraspinal germ cell tumors; Xb = Other and unspecified non-gonadal germ cell
tumors; Xc = Gonadal (ovarian and testicular) germ cell tumors; Xd = Gonadal carcinoma;
Xe - Other and unspecified malignant gonadal tumors.
63
59
82 82 82
87
70
72
94
93
94
89
Xa Xb Xc Male Female Xd Xe
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Xc
##

ICCC X
134
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
Table X.5: Current knowledge on causes of childhood malignant germ cell tumors (MGCT)
Exposure or Characteristic Comments
References
Cryptorchidism Risk is increased 2.5 - 11-fold. The contralateral as well
as ipsilateral testis is at increased risk.
34-36
High maternal hormone levels
during pregnancy
Use of oral contraceptives during pregnancy, high pre-
pregnancy weight, bleeding, hyperemesis and spotting
indicate high hormone levels.
34,35,37-39
Family history of germ cell
tumor
When malignant germ cell tumors occur in the same
family, they are usually of the same histologic type.
40,41
Hernia Central nervous system and genitourinary anomalies
have also been observed in germ cell tumor patients.
34,35,42
Pre-term birth Excess risk was not explained by cryptorchidism. 43,44
Trauma The causality of this association is not clear. Trauma
may result in closer scrutiny and earlier detection of an
existing tumor.
45-47
Virus infection, e.g., mumps,
cytomegalovirus, Epstein-B
virus, and parvovirus B19
48-52
High birth weight 35,43,44
Prenatal X-ray exposure 43,53
Parental occupation Associations have been observed with maternal
employment in the medical field, paternal employment
in service stations and aircraft industry, and paternal
exposure to x-rays, maternal exposure to solvents,
plastic and resin fumes.
44,54,55
Constitutional chromosome
abnormalities, particularly sex
chromosome abnormalities
(e.g., Klinefelter syndrome
(47,XXY), inverted Y)
56-60
Known risk factors
Factors for which evidence
is suggestive but not
conclusive
Factors for which evidence
is inconsistent or limtied

ICCC X
135
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
The increase in survival for this
subgroup, ICCC Xb, was dramatic
for children younger than 5 years of
age; the survival rate increased from
38% to 86%.
• The increase in survival between
1975-84 and 1985-94 was similar for
ovarian and testicular germ cell
tumors (Figure X.7). Both increased
from 82% to 93-94%.
The improvement in outcome observed in
the more recent period for children with
germ cell tumors likely represents the
widespread application of platinum-based
chemotherapy, which is particularly
effective against germ cell tumors [33].
RISK FACTORS
The etiology of malignant germ cell
tumors is poorly understood.
Cryptorchidism is the only confirmed risk
factor for testicular germ cell tumors (see
Table X.5 for references). Although rare,
testicular cancer coincidence in father and
son, and in male siblings has been
reported, implying a genetic contribution
in the disease origination. Suggested risk
factors for malignant germ cell tumors,
mainly based on findings from studies of
testicular cancer among adult
populations, include maternal exogenous
hormone use and high endogenous
hormone level during pregnancy, pre-term
birth, high birth weight, hernia, trauma,
pre-natal X-ray exposure, virus infection,
parental occupation and occupational
exposures, and certain constitutional
chromosome abnormalities.
SUMMARY
The ICCC Diagnostic Group X for
GCTOG tumors represents less than 4% of
tumors among children younger than 15
years of age. However, for the 15-19 year
age group, these tumors account for a much
higher proportion (approximately 16%) of
cancer cases. The age-incidence pattern for
the group of GCTOG tumors is character-
ized by relatively high rates in the first
year of life, followed by much lower rates
until puberty, when incidence begins to
increase and reaches rates greater than
those in the first year of life. For males, the
majority of testicular cancers occurring
before age 15 years are diagnosed in the
first 4 years of life. However, because the
incidence of testicular germ cell tumors
increases rapidly after age 15, the vast
majority of testicular cancer cases among
those younger than 20 years of age develop
among 15-19 year olds. Black males have a
much lower incidence of testicular germ cell
tumors than white males, while black
females and white females have similar
rates for ovarian germ cell tumors.
The distinctive nature of the germ cell
tumors of infants and young children
compared to those of adolescents and young
adults complicates analyses of trends in
incidence for the children younger than 20
years of age. However, over the past 20
years there has been a small absolute
increase in incidence for germ cell tumors
for children younger than age 15 years,
with most of the increase due to higher
rates for extragonadal germ cell tumors.
Among children younger than 20 years of
age, the incidence of GCTOG tumors has
increased. The increase has been primarily
driven by higher rates for gonadal germ cell
tumors among 15-19 year olds and by
higher rates for gonadal carcinomas among
15-19 year old females. The latter increase
is attributable to changes in reporting of
ovarian tumors during this time period,
specifically inclusion of borderline tumors of
the ovary. The increases in gonadal germ
cell tumors for adolescents 15-19 years of
age mirrors that observed for young adults
with germ cell tumors. The onset of higher
rates in males and females at the time of
puberty as well as results from epidemio-
logical studies suggest a contributory role

ICCC X
136
National Cancer Institute
SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
for hormonal influences, although the
nature of these influences remains to be
elucidated.
Reference List
1. Dehner LP: Gonadal and extragonadal germ cell
enoplasia of childhood. Hum Pathol 14?493-511,
1983.
2. Castleberry R, Cushing B, Perlman E, et al:
Germ cell tumors. In Principles and Practice of
Pediatric Oncology (Pizzo P, Poplack D, eds).
Philadelphia: Lippincott-Raven, 1997, pp 921-
945.
3. Pinkerton CR: Malignant germ cell tumours in
childhood. Eur J Cancer 33:895-901; discussion
901-2, 1997.
4. Kramarova E, Stiller CA: The international
classification of childhood cancer. Int J Cancer
68:759-65, 1996.
5. Riopel MA, Spellerberg A, Griffin CA, et al:
Genetic analysis of ovarian germ cell tumors by
comparative genomic hybridization. Cancer Res
58:3105-10, 1998.
6. Dal Cin P, Dei Tos AP, Qi H, et al: Immature
teratoma of the pineal gland with isochromo-
some 12p. Acta Neuropathol (Berl) 95:107-10,
1998.
7. Lemos JA, Barbieri-Neto J, Casartelli C:
Primary intracranial germ cell tumors without
an isochromosome 12p. Cancer Genet Cytogenet
100:124-8, 1998.
8. de Bruin TW, Slater RM, Defferrari R, et al:
Isochromosome 12p-positive pineal germ cell
tumor. Cancer Res 54:1542-4, 1994.
9. Shen V, Chaparro M, Choi BH, et al: Absence of
isochromosome 12p in a pineal region malignant
germ cell tumor. Cancer Genet Cytogenet
50:153-60, 1990.
10. Dal Cin P, Drochmans A, Moerman P, et al:
Isochromosome 12p in mediastinal germ cell
tumor. Cancer Genet Cytogenet 42:243-51, 1989.
11. Aly MS, Dal Cin P, Jiskoot P, et al: Competitive
in situ hybridization in a mediastinal germ cell
tumor. Cancer Genet Cytogenet 73:53-6, 1994.
12. Oosterhuis JW, Castedo SM, de Jong B, et al:
Ploidy of primary germ cell tumors of the testis.
Pathogenetic and clinical relevance. Lab Invest
60:14-21, 1989.
13. Silver SA, Wiley JM, Perlman EJ: DNA ploidy
analysis of pediatric germ cell tumors. Mod
Pathol 7:951-6, 1994.
14. Perlman EJ, Valentine MB, Griffin CA, et al:
Deletion of 1p36 in childhood endodermal sinus
tumors by two-color fluorescence in situ hybrid-
ization: a pediatric oncology group study. Genes
Chromosomes Cancer 16:15-20, 1996.
15. Perlman EJ, Cushing B, Hawkins E, et al:
Cytogenetic analysis of childhood endodermal
sinus tumors: a Pediatric Oncology Group study.
Pediatr Pathol 14:695-708, 1994.
16. Rodriguez E, Houldsworth J, Reuter VE, et al:
Molecular cytogenetic analysis of i(12p)-
negative human male germ cell tumors. Genes
Chromosomes Cancer 8:230-6, 1993.
17. Bosl GJ, Ilson DH, Rodriguez E, et al: Clinical
relevance of the i(12p) marker chromosome in
germ cell tumors. J Natl Cancer Inst 86:349-55,
1994.
18. Mostert MC, Verkerk AJ, van de Pol M, et al:
Identification of the critical region of 12p over-
representation in testicular germ cell tumors of
adolescents and adults. Oncogene 16:2617-27,
1998.
19. van Echten J, Oosterhuis JW, Looijenga LH, et
al: No recurrent structural abnormalities apart
from i(12p) in primary germ cell tumors of the
adult testis. Genes Chromosomes Cancer
14:133-44, 1995.
20. Tulinius H, Day N, Muir CS: Rarity of testis
cancer in Negroes. Lancet 1:35-6, 1973.
21. Spitz MR, Sider JG, Pollack ES, et al: Incidence
and descriptive features of testicular cancer
among United States whites, blacks, and
Hispanics, 1973-1982. Cancer 58:1785-90, 1986.
22. Brown LM, Pottern LM, Hoover RN, et al:
Testicular cancer in the United States: trends in
incidence and mortality. Int J Epidemiol 15:164-
70, 1986.
23. Hawkins EP: Germ cell tumors. Am J Clin
Pathol 109:S82-8, 1998.
24. Malogolowkin MH, Mahour GH, Krailo M, et al:
Germ cell tumors in infancy and childhood: a
45-year experience. Pediatr Pathol 10:231-41,
1990.
25. Marsden HB, Birch JM, Swindell R: Germ cell
tumours of childhood: a review of 137 cases. J
Clin Pathol 34:879-83, 1981.
26. Hawkins E, Issacs H, Cushing B, et al: Occult
malignancy in neonatal sacrococcygeal terato-
mas. A report from a Combined Pediatric
Oncology Group and Children’s Cancer Group
study. Am J Pediatr Hematol Oncol 15:406-9,
1993.
27. Gilcrease MZ, Brandt ML, Hawkins EP: Yolk
sac tumor identified at autopsy after surgical
excision of immature sacrococcygeal teratoma. J
Pediatr Surg 30:875-7, 1995.
28. Bergstrom R, Adami HO, Mohner M, et al:
Increase in testicular cancer incidence in six
European countries: a birth cohort phenomenon.
J Natl Cancer Inst 88:727-33, 1996.
29. Stone JM, Cruickshank DG, Sandeman TF, et
al: Trebling of the incidence of testicular cancer
in victoria, Australia (1950-1985). Cancer
68:211-9, 1991.
30. Pearce N, Sheppard RA, Howard JK, et al: Time
trends and occupational differences in cancer of

ICCC X
137
National Cancer Institute SEER Pediatric Monograph
GERM CELL, TROPHOBLASTIC AND OTHER GONADAL NEOPLASMS
the testis in New Zealand. Cancer 59:1677-82,
1987.
31. Walker AH, Ross RK, Pike MC, et al: A possible
rising incidence of malignant germ cell tumours
in young women. Br J Cancer 49:669-72, 1984.
32. dos Santos Silva I, Swerdlow AJ: Ovarian germ
cell malignancies in England: epidemiological
parallels with testicular cancer. Br J Cancer
63:814-8, 1991.
33. Mann JR, Raafat F, Robinson K, et al:
UKCCSG’s germ cell tumour (GCT) studies:
improving outcome for children with malignant
extracranial non-gonadal tumours—carboplatin,
etoposide, and bleomycin are effective and less
toxic than previous regimens. United Kingdom
Children’s Cancer Study Group. Med Pediatr
Oncol 30:217-27, 1998.
34. Depue RH, Pike MC, Henderson BE: Estrogen
exposure during gestation and risk of testicular
cancer. J Natl Cancer Inst 71:1151-5, 1983.
35. Henderson BE, Benton B, Jing J, et al: Risk
factors for cancer of the testis in young men. Int
J Cancer 23:598-602, 1979.
36. Strader CH, Weiss NS, Daling JR, et al: Cryp-
torchism, orchiopexy, and the risk of testicular
cancer. Am J Epidemiol 127:1013-8, 1988.
37. Swerdlow AJ, Stiller CA, Wilson LM: Prenatal
factors in the aetiology of testicular cancer: an
epidemiological study of childhood testicular
cancer deaths in Great Britain, 1953-73. J
Epidemiol Community Health 36:96-101, 1982.
38. Walker AH, Ross RK, Haile RW, et al: Hormonal
factors and risk of ovarian germ cell cancer in
young women. Br J Cancer 57:418-22, 1988.
39. Schottenfeld D, Warshauer ME, Sherlock S, et
al: The epidemiology of testicular cancer in
young adults. Am J Epidemiol 112:232-46, 1980.
40. Klein FA, Pollack MS, Sogani PC, et al: Father-
son testicular (teratocarcinoma) malignancy.
Urology 22:281-3, 1983.
41. Dieckmann KP, Becker T, Jonas D, et al:
Inheritance and testicular cancer. Arguments
based on a report of 3 cases and a review of the
literature. Oncology 44:367-77, 1987.
42. Morrison AS: Cryptorchidism, hernia, and
cancer of the testis. J Natl Cancer Inst 56:731-3,
1976.
43. Brown LM, Pottern LM, Hoover RN: Prenatal
and perinatal risk factors for testicular cancer.
Cancer Res 46:4812-6, 1986.
44. Shu XO, Nesbit ME, Buckley JD, et al: An
exploratory analysis of risk factors for childhood
malignant germ-cell tumors: report from the
Childrens Cancer Group (Canada, United
States). Cancer Causes Control 6:187-98, 1995.
45. Haughey BP, Graham S, Brasure J, et al: The
epidemiology of testicular cancer in upstate
New York. Am J Epidemiol 130:25-36, 1989.
46. Swerdlow AJ, Huttly SR, Smith PG: Testicular
cancer and antecedent diseases. Br J Cancer
55:97-103, 1987.
47. Strader CH, Weiss NS, Daling JR: Vasectomy
and the incidence of testicular cancer. Am J
Epidemiol 128:56-63, 1988.
48. Mueller N, Hinkula J, Wahren B: Elevated
antibody titers against cytomegalovirus among
patients with testicular cancer. Int J Cancer
41:399-403, 1988.
49. Heinzer H, Dieckmann KP, Huland E: Virus-
related serology and in situ hybridization for
the detection of virus DNA among patients with
testicular cancer. Eur Urol 24:271-6, 1993.
50. Birch JM, Marsden HB, Swindell R: Pre-natal
factors in the origin of germ cell tumours of
childhood. Carcinogenesis 3:75-80, 1982.
51. Oliver RT: Atrophy, hormones, genes and
viruses in aetiology germ cell tumours. Cancer
Surv 9:263-86, 1990.
52. Gray A, Guillou L, Zufferey J, et al: Persistence
of parvovirus B19 DNA in testis of patients
with testicular germ cell tumours. J Gen Virol
79:573-9, 1998.
53. Loughlin JE, Robboy SJ, Morrison AS: Risk
factors for cancer of the testis [letter]. N Engl J
Med 303:112-3, 1980.
54. Kardaun JW, Hayes RB, Pottern LM, et al:
Testicular cancer in young men and parental
occupational exposure. Am J Ind Med 20:219-27,
1991.
55. Johnston HE, Mann JR, Williams J, et al: The
Inter-Regional, Epidemiological Study of
Childhood Cancer (IRESCC): case-control study
in children with germ cell tumours. Carcinogen-
esis 7:717-22, 1986.
56. Dmitrovsky E, Bosl GJ, Chaganti RS: Clinical
and genetic features of human germ cell cancer.
Cancer Surv 9:369-86, 1990.
57. Surti U, Hoffner L, Chakravarti A, et al: Genet-
ics and biology of human ovarian teratomas. I.
Cytogenetic analysis and mechanism of origin.
Am J Hum Genet 47:635-43, 1990.
58. Teter J, Boczkowski K: Occurrence of tumors in
dysgenetic gonads. Cancer 20:1301-10, 1967.
59. Atkin NB, Baker MC: X-chromatin, sex chromo-
somes, and ploidy in 37 germ cell tumors of the
testis. Cancer Genet Cytogenet 59:54-6, 1992.
60. Hasle H, Mellemgaard A, Nielsen J, et al:
Cancer incidence in men with Klinefelter
syndrome. Br J Cancer 71:416-20, 1995.

138
National Cancer Institute
SEER Pediatric Monograph

ICCC XI
139
National Cancer Institute
SEER Pediatric Monograph
CARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
Leslie Bernstein, James G. Gurney
HIGHLIGHTS
Incidence
♦ Among children, particularly before the adolescent years, carcinomas are very rare.
♦ In the US, approximately 1,050 children and adolescents younger than 20 years of
age are diagnosed with carcinomas each year, of which approximately 350 are
thyroid carcinomas and 300-350 are melanomas.
♦ All of the carcinomas combined comprised 9.2% of cancer in children younger than
20. The majority of the carcinomas were either thyroid carcinomas (35.5%) or
melanomas (30.9%). Adrenocortical carcinomas (1.3%), nasopharyngeal carcinomas
(4.5%), and other skin carcinomas (0.5%) combined for only a small proportion of
the total, while other and unspecified carcinomas comprised 27.3%.
♦ The incidence rates for thyroid carcinoma were highest among the 15-19 year olds
and much higher among females (24.4 per million) than males (4.7 per million)
(Table XI.2).
♦ The incidence rates for malignant melanoma were highest among the 15-19 year
olds and higher among females (16.5 per million) than males (10.0 per million)
(Table XI.2).
Survival
♦ The 5-year survival rate was 99% for thyroid carcinomas. Males had a slightly
lower survival rate than females (Table XI.4).
♦ The 5-year survival rate was 91% for malignant melanoma. Females had a 93%
survival rate compared to males with an 87% survival rate (Table XI.4).
Risk factors
♦ The most well established risk factor for thyroid carcinoma is ionizing radiation
exposure, from both environmental and therapeutic sources.
♦ The primary risk factors for melanoma are sun exposure and number of melanocytic
and dysplastic nevi.
INTRODUCTION
Carcinomas are malignancies that
originate in epithelial tissues. Epithelial
cells cover the external surface of the body,
line the internal cavities, and form the
lining of glandular tissues [1,2]. Cancers
that originate from epithelial cells, includ-
ing those of the breast, lung, prostate, and
colon, are by far the most common types of
cancer in adults. Among children, however,
particularly before the adolescent years,
carcinomas are very rare. Leukemias,
central nervous system cancer, lymphomas,
sarcomas, and the embryonal cancers such
as neuroblastoma, retinoblastoma and
Wilms’ tumor, represent a far greater
burden to young children than does any
specific epithelial cancer. Nevertheless, a
variety of carcinomas do occur in children,
especially during late adolescence, and in
this report we provide descriptive epidemio-
logic data to characterize their occurrence.
In the US, approximately 1,050 children
and adolescents younger than 20 years of
age are diagnosed with carcinomas each
year, of which approximately 350 are
thyroid carcinomas and 300-350 are mela-
nomas.

ICCC XI CARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
140
National Cancer Institute
SEER Pediatric Monograph
Classification system
Although other classification systems
exist [2], the diverse types of epithelial
malignancies that are called ‘carcinomas
and other malignant epithelial neoplasms’
by the ICCC system are classified into six
broad subgroups [3]:
a. adrenocortical carcinoma
b. thyroid carcinoma
c. nasopharyngeal carcinoma
d. malignant melanoma
e. skin carcinoma other than mela-
noma
f. other and unspecified carcinomas
All of the malignancies within the group
‘other and unspecified carcinomas’ are very
rare in children. Neoplasms of the salivary
gland, colon, appendix, lung and bronchus,
uterine cervix, and urinary bladder account
for most of this group. Likewise, the
adrenocortical, nasopharyngeal, and skin
carcinomas (other than melanoma) rarely
occur in children. Because thyroid carcino-
mas and malignant melanoma (henceforth
called melanoma) are the only epithelial
malignancies that occur with any signifi-
cant frequency in children, we will focus
primarily on these two cancers in this
report. It should be noted that the ICCC
system includes germ cell carcinomas
within ‘germ cell, trophoblastic, and other
gonadal neoplasms’, rather than ‘carcino-
mas and other malignant epithelial neo-
plasms’. The incidence rate of gonadal
carcinomas for children younger than 20
years of age is only 1.0 per million, so this
omission will not appreciably influence our
results. Please note also that we have
calculated frequencies and rates from
SEER data for the years 1975 through
1995, and, unless otherwise specified, we
report incidence rates as average annual
rates per million children younger than 20
years of age, adjusted to the 1970 US
standard population.
Thyroid carcinomas are endocrine
tumors, although they do not necessarily
exhibit hormonal activity. There are four
types of thyroid carcinomas: papillary,
follicular, medullary and anaplastic. In
children, papillary tumors represent
greater than 70% of thyroid carcinomas,
and follicular tumors another 20%. Only a
small proportion of thyroid carcinomas are
medullary (5-10%), or anaplastic (extremely
rare). The vast majority of childhood
thyroid carcinomas are well differentiated
tumors, and despite their malignant pathol-
ogy, the clinical course of most thyroid
carcinomas is relatively benign [4-6].
Malignant cutaneous melanomas arise
from epidermal melanocytes and are often
classified into one of three histopathological
types according to the presence and pattern
of intraepidermal growth: superficial
spreading melanoma, nodular melanoma,
and lentigo maligna melanoma. Melano-
mas occur most frequently on the trunk in
white males, the lower limbs in white
females, and the soles of the feet in blacks
[7,8].
INCIDENCE
All of the carcinomas combined com-
prised 9.2% of cancer in children during the
time period of this study. The majority of
the 2,735 epithelial cancers were either
thyroid carcinomas (35.5%) or melanomas
(30.9%). Adrenocortical carcinomas (1.3%),
nasopharyngeal carcinomas (4.5%), and
other skin carcinomas (0.5%) combined for
only a small proportion of the total, while
other and unspecified carcinomas com-
prised 27.3%. Nearly 75% (2,047) of the
childhood carcinomas occurred in adoles-
cents 15-19 years of age, including 75% of
the thyroid carcinomas, 80% of the melano-
mas, 63% of the nasopharyngeal carcino-
mas, and 74% of the other and unspecified
carcinomas. Although there were only 36
adrenocortical carcinomas reported to

ICCC XICARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
141
National Cancer Institute SEER Pediatric Monograph
SEER areas, 18 of them occurred in chil-
dren younger than 5 years of age.
Age-specific incidence
The impressive age differences in inci-
dence rates of carcinomas are shown in
Table XI.1. Incidence rates of thyroid
carcinoma and melanoma were practically
negligible in very young children. Among
15-19 year olds, however, both melanoma
and thyroid cancer substantially increased
in occurrence over younger ages, particu-
larly for females. The male-to-female ratios
Table XI.1: Average annual age-specific incidence rates per million by histology and
sex, all races, SEER, 1975-95
Tumor type Age Total Females Males F/M Ratio
All carcinomas 0-4 1.6 1.9 1.3 1.5
5-9 2.7 3.1 2.3 1.3
10-14 10.0 12.1 8.0 1.5
15-19 40.3 55.5 25.9 2.1
Adrenocortical carcinoma 0-4 0.4 0.5 0.3 1.7
5-9 0.1 0.1 0
10-14 0.1 0.1 0.1 1.0
15-19 0.2 0.2 0.1 2.0
Thyroid carcinoma 0-4 0.1 0.2 0
5-9 1.0 1.3 0.8 1.6
10-14 3.9 6.0 1.8 3.3
15-19 14.4 24.4 4.7 5.2
Nasopharyngeal carcinoma 0-4 0.1 0 0.1
5-9 0.1 0 0.1
10-14 0.8 0.3 1.30 0.2
15-19 1.5 1.2 1.8 0.7
Malignant melanoma 0-4 0.7 0.7 0.7 1.0
5-9 0.7 0.8 0.7 1.1
10-14 2.2 2.5 1.9 1.3
15-19 13.2 16.5 10.0 1.7
Skin carcinoma other than melanoma 0-4 0 0 0
5-9 0 0 0
10-14 0.1 0.1 0.1 1.0
15-19 0.2 0.2 0.1 2.0
Other and unspecified carcinomas 0-4 0.4 0.5 0.3 1.7
5-9 0.8 0.9 0.6 1.5
10-14 2.8 3.0 2.7 1.1
15-19 11.0 12.8 9.2 1.4

ICCC XI CARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
142
National Cancer Institute
SEER Pediatric Monograph
in rates were greatest among adolescents
15-19 years, and the sex difference was
most pronounced for thyroid carcinoma.
Figure XI.1 provides greater detail on the
age-specific patterns of incidence for all
carcinomas combined
1
. Carcinoma inci-
dence was quite low through age 10 years,
but the rates increased dramatically in
older children. At age 19, incidence rates
were 35 per million males and 75 per
million females. For thyroid carcinoma
(Figure XI.2), the age-specific incidence
rates for males and females began to
diverge at age 10 years. Beginning at age
13, the rates increased substantially for
females, while the increase in male rates
was more modest. Incidence rates of mela-
noma are shown in Figure XI.3. Rates
differed minimally by sex until age 16,
when the rates for females became greater
than those for males.
Sex-specific and race-specific incidence
As shown in Table XI.2, the incidence
rate of all childhood carcinomas combined
was 13.8 per million. The ratio of white to
black rates was 1.5 to 1. The magnitude of
this racial difference is explained by the 2.5
to 1 ratio for thyroid cancer and the dra-
Figure XI.1: Total carcinoma age-specific incidence rates
by sex, all races, SEER, 1976-84 and 1986-94
)
)
)
)
)
)
)
)
))
)
)
)
)
)
)
)
)
)
)
"
"
"
"
""
"
"
"
"
"
"
"
""
"
"
"
"
"
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
40
50
60
70
80
Average annual rate per million
Male
Female
"
)
1
Enumeration of the population at risk by single years of age was
available only for the census years 1980 and 1990. The US Bureau
of the Census provides intercensal population estimates by 5-year
age groups, but not by single years of age. Therefore, the
population estimates for 1980 were used in rate calculations for
cases diagnosed from 1976-84 and the 1990 estimates were used for
cases diagnosed from 1986-94.

ICCC XICARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
143
National Cancer Institute SEER Pediatric Monograph
Figure XI.2: Thyroid carcinoma age-specific
incidence rates, by sex, all races
SEER, 1976-84 and 1986-94
)
)
))
)
))
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"""""
"
"
"
"
"
"
"
"
"
"
"
"
"
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 1617 18 19 20
Age (in years) at diagnosis
0
10
20
30
40
Average annual rate per million
Male
Female
"
)
Figure XI.3: Malignant melanoma age-specific
incidence rates, by sex, all races
SEER, 1976-84 and 1986-94
)
)
)
)
)))
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
""
""
"
"
"
"
"
"
"
"
"
"
"
"
"
"
01234567891011121314151617181920
Age (in years) at diagnosis
0
10
20
30
Average annual rate per million
Male
Female
"
)
matic 16 to 1 ratio for melanoma. The
variation in carcinoma rates between
whites and blacks was greater among
females than males.
Table XI.2 further illustrates that
incidence rates of thyroid carcinoma were
over 4-fold higher in females than males for
both white and black children. Among
whites, melanoma was more common in
females than males, but not nearly to the
extent that was observed for thyroid cancer.
In fact, among whites, male rates of mela-
noma were higher than male rates of
thyroid cancer. As is well known, occur-
rence of melanoma in blacks is extremely
unusual. Although not shown in the table,
black children had a slightly higher rate of
other and unspecified carcinomas than
white children (4.7 versus 3.6 per million,
respectively).
Table XI.2: Average annual age-adjusted* incidence rates per million
by histology, race, and sex, age <20, SEER, 1975-95
Tumor type Race Total Females Males F/M Ratio
All carcinomas All races 13.8 18.2 9.5 1.9
Whites 14.2 19.3 9.4 2.1
Blacks 9.3 10.7 7.8 1.4
Thyroid carcinoma All races 4.9 8.1 1.9 4.3
Whites 5.2 8.6 2.0 4.3
Blacks 2.1 3.4 0.7 4.9
Malignant melanoma All races 4.2 5.1 3.3 1.5
Whites 4.8 5.8 3.8 1.5
Blacks 0.3 0.2 0.3 0.7
* Adjusted to the 1970 US standard population

ICCC XI CARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
144
National Cancer Institute
SEER Pediatric Monograph
TRENDS
Figure XI.4 illustrates yearly rates of
carcinomas from 1975-95. Age-adjusted
incidence rates did not increase appreciably
over this time frame for either melanoma
or thyroid carcinoma. Incidence rates over
time are further illustrated in Table XI.3,
where rates by sex are shown within spe-
cific time periods.
SURVIVAL
Survival probability is excellent for
children with either thyroid carcinoma or
melanoma. Table XI.4 provides 5-year
relative survival probabilities for both
major types of childhood carcinoma, as well
as for all carcinomas combined. Virtually
no change in survival was observed for
thyroid carcinoma over the time periods
1975-84 versus 1985-94 (Figure XI.5).
Survival improved slightly for melanoma,
from 85% for those diagnosed during 1975-
84, to 91% during 1985-94 (Figure XI.6).
Figure XI.4: Trends in carcinoma age-adjusted*
incidence rates, by histology, age <20
all races, both sexes, SEER 1975-95
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
+
+
+
+
+
+++
+
+
+
+
+
+
+
+
+
+
+
+
+
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
75 77 79 81 83 85 87 89 91 93 95
Year of diagnosis
0
2
4
6
8
10
12
14
16
18
20
Average annual rate per million
All carcinomas
Thyroid carcinoma
Malignant melanoma
#
+
(
*Adjusted to the 1970 US standard population
Table XI.3: Trends in average annual age-adjusted* incidence rates
per million, by histology and sex, age <20, all races
SEER, 1975-95
Tumor type Year of diagnosis Total Females Males
All carcinomas 1975-79 13.6 18.7 8.7
1980-84 12.9 16.1 9.4
1985-89 14.1 18.4 10.1
1990-95 14.3 19.3 9.7
Thyroid carcinoma 1975-79 4.8 7.8 1.8
1980-84 4.7 7.2 2.4
1985-89 5.2 8.8 1.8
1990-95 4.9 8.6 1.5
Malignant melanoma 1975-79 4.1 5.1 3.1
1980-84 3.6 4.4 2.8
1985-89 4.6 5.5 3.9
1990-95 4.6 5.6 3.6
* Adjusted to the 1970 US standard population

ICCC XICARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
145
National Cancer Institute SEER Pediatric Monograph
For each type of carcinoma shown, females
had slightly better survival probabilities
than males. Black children appeared to
fare worse than white children for all
carcinomas combined, however there were
only 29 black males and 34 black females
during the time period, so the results
should be viewed very cautiously. Simi-
larly, there were too few black children with
either thyroid carcinoma or melanoma on
whom to base relative survival rates.
RISK FACTORS
Risk factors for thyroid carcinoma have
been reviewed in several previous reports
[9-15] and will only be briefly summarized
here. The most well established risk factor
for thyroid carcinoma is ionizing radiation
exposure, from both environmental and
therapeutic sources. Irradiation treatment
for conditions such as tinea capitis, en-
larged thymus, acne, and cancer have
clearly been shown to increase risk for
thyroid carcinoma development. Other
than cancer treatment, however, these
causes are primarily of historical concern.
Environmental exposures to ionizing radia-
tion from the atomic bombings in Japan
and from the nuclear disaster at Chernobyl
also have definitively been shown to cause
substantial increases in thyroid carcinoma.
The preponderance of thyroid cancer in
females suggests that hormonal factors
may mediate disease occurrence. Other
potential etiologic factors include benign
thyroid diseases and certain inherited
cancer susceptibility syndromes, such as
familial adenomatous polyposis, and mul-
tiple endocrine neoplasia (MEN) types I,
IIA and IIB.
The primary risk factors for melanoma
are sun exposure and number of
melanocytic and dysplastic nevi. An exten-
sive review of studies related to sun expo-
Table XI.4: Five-year relative survival rates by histology, race and sex
age <20, SEER, 1985-94
Tumor type Total Females Males
Percent Percent Percent
All carcinomas
All races 89 93 83
White 91 93 86
Black 77 85 65
Thyroid carcinoma
All races 99 99 95
White 98 99 94
Black * * *
Malignant melanoma
All races 91 93 87
White 91 93 88
Black * * *
*Less than 25 cases.

ICCC XI CARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
146
National Cancer Institute
SEER Pediatric Monograph
Figure XI.5: Thyroid carcinoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
# - <25 cases - rate not shown
99 99 99 99
100
9999
95
99
98 98
99
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
#
#
#
Figure XI.6: Malignant melanoma 5-year relative survival rates
by sex, race, age and time period, SEER (9 areas), 1975-84 and 1985-94
# - <25 cases - rate not shown
85
80
88
85
94
84
91
87
93
91
86
92
Total Male Female White Black <5 5-9 10-14 15-19
0
20
40
60
80
100
Percent surviving 5 years
1975-84
1985-94
Sex
Race
Age
###

ICCC XICARCINOMAS AND OTHER MALIGNANT EPITHELIAL NEOPLASMS
147
National Cancer Institute SEER Pediatric Monograph
sure, nevi, and other potential risk factors
can be found in reference 6.
SUMMARY
In stark contrast to cancer incidence in
adults, carcinomas were very rare in chil-
dren, especially those younger than 15
years of age. Rates increased quite sub-
stantially with increasing age, however,
particularly among females aged 10-19
years. The most common types of epithelial
cancer in children were thyroid carcinomas
(4.9 per million younger than 20 years of
age) and melanomas (4.2 per million
younger than 20 years of age). Females,
however, had 4 times the thyroid cancer
rate observed for males. Additionally, white
children had 2.5 times the thyroid cancer
rate and 16 times the melanoma rate
observed for black children. From 1975-95
incidence rates of both thyroid cancer and
melanoma remained fairly stable. The
strongest known risk factor for thyroid
carcinoma is ionizing radiation exposure.
Sun exposure and number of nevi are the
best described risk factors for melanoma
occurrence. Fortunately, 5-year survival
probability is excellent for both thyroid
cancer (99%) and melanoma (91%).
Reference List
1. Junqueira LC, Carneiro J, Kelly, RO. Basic
Histology, 7
th
Edition. Appleton & Lange,
Norwalk, CT, 1992; 66-94.
2. Berg JW. Morphologic classification of human
cancer. In: Shottenfeld D, Fraumeni JF (eds):
Cancer Epidemiology and Prevention, 2
nd
edition. New York: Oxford University Press,
1996; 28-44.
3. Kramarova E, Stiller CA. The international
classification of childhood cancer. Int J Cancer.
1996;68:759-765.
4. Stratakis CA, Chrousos GP. Endocrine tumors.
In: Pizzo PA, Poplack DG, Editors. Principles
and Practices of Pediatric Oncology. 3rd ed.
Philadelphia, PA: Lippencott-Raven; 1997: 947-
976.
5. Danese D, Gardini A, Farsetti A, Sciacchitano S,
Andreoli M, Ponecorvi A. Thyroid carcinoma in
children and adolescents. Eur J Pediatr; 1997;
156:190-194.
6. McClollan DR, Francis GL. Thyroid cancer in
children, pregnant women, and patients with
Graves’ disease. Endocrin Metab Clin North
Am, 1996; 25:27-47.
7. Armstrong BK, English DR. Cutaneous malig-
nant melanoma. In: Schottenfeld D, Fraumeni
JF, Editors. Cancer Epidemiology and Preven-
tion. 2nd ed. New York: Oxford University
Press; 1996; 1282-1312.
8. Armstrong BK, Kricker A.. Skin Cancer.
Dermatologic Clin. 1995; 13:583-594.
9. Ron E. Thyroid Cancer. In: Schottenfeld D,
Fraumeni JF, Editors. Cancer Epidemiology and
Prevention. 2nd ed. New York: Oxford Univer-
sity Press; 1996; 1000-1021.
10. Nikiforov YE, Fagin JA. Risk factors for thyroid
cancer. Trends Endocrinol Metab, 1997: 8:20-25.
11. Fraker DL. Radiation exposure and other
factors that predispose to human thyroid
neoplasia. Surg Clin North Am, 1995; 3:365-375.
12. Sokic SI, Adanja BJ, Vlajinac HD, Jankovic RR,
Marinkovic JP, Zivaljevic VR. Risk factors for
thyroid cancer. Neoplasma, 1994; 41:371-374.
13. Akslen LA. Thyroid cancer: some aspects of
epidemiology and etiological factors, pathologi-
cal features and tumour biology (review). Int J
Onc 1994; 4:931-942.
14. Geirger JD, Thompson NW. Thyroid tumors in
children. Otolaryngologic Clinics of North
America. 1996; 29:711-719.
15. Tucker MA, Morris Jones PH, Boice JD, Robison
LL, Stone BJ, Stovall M, Jenkin RDT, Lubin JH,
Baum ES, Siegel SE, Meadows AT, Hoover RN,
Fraumeni JF. Therapeutic radiation at a young
age is linked to secondary thyroid cancer.
Cancer Res, 1991; 51:2885-2888.

148
National Cancer Institute
SEER Pediatric Monograph

XII
149
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG INFANTS
HIGHLIGHTS
Incidence
♦ The age of peak cancer incidence among children occurred during the first year of
life (Figure XII.1).
♦ Malignancies of infancy represented 10% of all cancer that was diagnosed among
children younger than 15 years of age. The average annual incidence rate of all
infant cancer combined was 233 per million infants, which was 12% higher than the
age (2 years) with the next highest incidence.
♦ The rate among females (234 per million infants) was essentially the same as that
in males (232 per million infants). This is notable because infancy was the only age
among children younger than 15 years of age in which female rates were not lower
than male rates.
♦ Neuroblastoma comprised 28% of infant cancer cases and was the most common
malignancy among these young children (65 per million infants).
♦ The leukemias as a group (41 per million infants) represented the next most com-
mon type of cancer, comprising 17% of all cases (Figure XII.2).
♦ Central nervous system malignancies comprised 13% of infant cancer, with an
average annual incidence rate of nearly 30 per million infants.
♦ The average annual incidence rates for malignant germ cell and malignant soft
tissue tumors were essentially the same at 15 per million infants. Each comprised
about 6% of infant cancer (Figure XII.2).
♦ Leukemias accounted for a substantial proportion of the racial difference, in that the
average annual rate for white infants (48.7 per million) was 66% higher than for
black infants (29.4 per million).
Survival
♦ The prognosis for infants with cancer is often worse than in children of older ages,
even when comparing the same histologic diagnosis. For instance, the 5-year rela-
tive survival for children younger than 15 years of age who were diagnosed with
acute lymphoid leukemia from 1975-94 was well over 70%, but for infants the sur-
vival rate was 33%.
♦ Over 80% of children diagnosed with neuroblastoma during infancy were alive 5
years following diagnosis. In contrast, for children diagnosed with neuroblastoma at
age 1 year or older, the 5-year relative survival was about 45%.
INTRODUCTION
Adult cancers usually form in epithelial
tissues and are believed often to be the
result of a long biological process related to
the interaction of exogenous exposures with
genetic and other endogenous characteris-
tics among susceptible people. However, in
young children, particularly infants, the
aberrant genetic processes that fail to
safeguard against the clonal proliferation of
cells with unregulated growth potential
occur very early in life and progress very
quickly. Due to the unique clinical, genetic,
and epidemiologic characteristics of cancers
in infants [1,2], it is becoming increasingly
James G. Gurney, Malcolm A. Smith, Julie A. Ross

XII
150
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG INFANTS
apparent that the study of infant cancer
may lead to further understanding of the
mechanisms of carcinogenesis. With that in
mind, this chapter will briefly summarize
and discuss data on cancer that occurs
among children who are diagnosed within
the first year of life.
INCIDENCE
The cancer cases used to calculate
incidence rates in this discussion were
limited to primary malignancies that were
registered in SEER areas of the United
States during the time periods 1976-84 and
1986-94. This time restriction was imposed
because enumeration of the population at
risk by single years of age was available
only for the census years of 1980 and 1990.
The US Bureau of the Census provides
intercensal population estimates by 5-year
age groups, but not by single years of age.
Therefore, the population estimates for
1980 were used in rate calculations for
cases diagnosed from 1976-84, and the
1990 estimates were used for cases
diagnosed from 1986-94.
The age of peak cancer incidence
among children occurred during the first
year of life, as shown in Figure XII.1.
Malignancies of infancy represented 10% of
all cancer that was diagnosed among
children younger than 15 years of age. The
incidence rate of all infant cancer combined
was 233 per million infants, which was 12%
higher than the age (2 years) with the next
highest incidence. The rate among females
(234 per million infants) was essentially the
same as that in males (232 per million
infants). This is notable because infancy
was the only age among children younger
than 15 years of age in which female rates
were not lower than male rates. Differ-
ences in rates by sex will be discussed in
more detail below.
Figure XII.1: Total childhood cancer age-specific incidence rates
by sex, all races, SEER, 1976-84 and 1986-94 combined
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
"
"
"
"
"
"
"
"
"
"
"
"
"
"
"
0123456789101112131415
Age (in years) at diagnosis
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
Average annual rate per million
Males
Females
Both sexes
"
)
#

XII
151
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG INFANTS
Histology-specific incidence
Figure XII.2 illustrates the incidence
rate for the most predominant types of
infant cancer. Although neuroblastoma
represented less than 8% of cancer cases
among children younger than 15 years of
age, neuroblastoma comprised 28% of
infant cancer cases and was the most
common malignancy among these young
children (65 per million infants). The
leukemias as a group (41 per million in-
fants) represented the next most common
type of cancer, comprising 17% of all cases.
As with older children, acute lymphoid
leukemia was the most frequently occur-
ring leukemia. The average annual inci-
dence rate of acute lymphoid leukemia was
21 per million infants, while the rate for the
acute non-lymphoid leukemias was 11 per
million infants. For juvenile chronic my-
eloid leukemia, which by international
consensus is now called juvenile
myelomonocytic leukemia (JMML), the
average annual incidence rate was 3 per
million infants. The combined rate for
other and unspecified leukemias was about
5 per million infants.
Central nervous system malignancies
comprised 13% of infant cancer, with an
incidence rate of nearly 30 per million
infants. Astrocytomas and other gliomas
(combined) accounted for half of the CNS
malignancies (15 per million), followed by
primitive neuroectodermal tumors/medullo-
blastomas (PNET, 9 per million infants)
and ependymomas (5 per million). Retino-
blastoma and Wilms’ tumor followed CNS
cancer in order of occurrence among in-
fants. Retinoblastoma accounted for about
12% of infant cancer and Wilms’ tumor an
additional 9%.
The incidence rates for malignant germ
cell tumors (including intracranial) and
malignant soft tissue tumors were essen-
tially the same at 15 per million infants.
64.6
40.5
29.7
26.7
22.5
15.3
15.2
9.5
4.4
2.8
1.2
0.5
Neuroblastoma
Leukemias
CNS
Retinoblastoma
Wilms'
Germ cell
Soft tissue
Hepatic
Lymphomas
Epithelial
Other/unspecified
Bone
0 10203040506070
Average annual rate per million
Figure XII.2: Infant age-specific incidence rates by type, all races,
both sexes, SEER, 1976-84 and 1986-94 combined

XII
152
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG INFANTS
Each comprised about 6% of infant cancer.
About 29% of the germ cell malignancies
diagnosed during infancy were gonadal,
which is half the percentage that occurs
overall in children younger than 15 years of
age. Unlike soft tissue tumors in older
children, the rate of fibrosarcomas (5 per
million infants) was similar to that of
rhabdomyosarcomas (6 per million infants).
This contrasts with the rhabdomyosarcoma
and fibrosarcoma rates among children
younger than 15 years of age of 4.6 per
million and 2.3 per million, respectively.
The rate for malignancies of the liver,
almost exclusively hepatoblastoma, was 9.5
per million infants. Hepatoblastoma is
similar to neuroblastoma, retinoblastoma,
and Wilms’ tumor (nephroblastoma) in that
it is an embryonal malignancy with the age
of peak incidence occurring during very
early childhood. Lymphomas and espe-
cially bone cancers, which are quite impor-
tant cancers among adolescents, are ex-
tremely rare in infants.
Sex-specific incidence
The female to male ratios of incidence
rates for selected cancer types are illus-
trated in Figure XII.3. None of the sex
differences were very pronounced. For
leukemias and CNS cancer, however, the
sex ratios differed by histologic subtype.
Figure XII.4 provides female to male ratios
for major subgroups of leukemia and CNS
cancer. This illustration reveals that most
types of infant leukemia and CNS cancer
were more common in females than males,
but JMML and ependymoma were notable
exceptions. For both types of neoplasms,
there is around a 2-fold higher average
annual incidence in males than in females.
The JMML rates were 4.0 per million male
infants compared with 1.7 per million
female infants. The ependymoma rates
0.86
1.17
1.07
1.19
1.1
0.92
0.91
0.8
Neuroblastoma
Leukemias
CNS
Retinoblastoma
Wilms'
Germ cell
Soft tissue
Hepatic
0 0.5 1 1.5 2
Ratio of female to male incidence rates
Figure XII.3: Ratios of female to male cancer incidence
rates among infants by type, all races
SEER, 1976-84 and 1986-94 combined
Figure XII.4: Ratios of female to male cancer incidence
rates among infants by type, all races
SEER, 1976-84 and 1986-94 combined
1.17
1.4
1.26
0.43
0.8
1.07
1.25
1.27
0.56
0.63
LEUKEMIAS
Acute lymphoid
Acute non-lymphoid
JMML
Other/Unspecified
CNS
Astrocytomas
PNET
Ependymoma
Other/Unspecified
0 0.5 1 1.5 2
Ratio of female to male incidence rates

XII
153
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG INFANTS
were 6.2 per million male infants compared
with 3.5 per million female infants.
Black-white differences in incidence
Figure XII.5 demonstrates the sub-
stantial discrepancy in black-white cancer
incidence rates among children, especially
young children. During infancy this racial
variation is quite pronounced, in that white
children (275 per million white infants) had
a 40% higher malignancy rate than black
children (196 per million black infants).
This difference does not appear to be a
result of earlier diagnosis for white chil-
dren, based on the similar age pattern of
incidence that is illustrated in Figure XII.5.
The ratios of incidence rates for white
relative to black infants are shown for
selected cancer types in Figure XII.6.
Leukemias accounted for a substantial
proportion of the racial difference, in that
the average annual rate for white infants
(48.7 per million) was 66% higher than for
black infants (29.4 per million). For neuro-
blastoma, white rates were 78% higher
than black rates (79.0 vs. 44.5 per million,
respectively). The relative difference was
even greater for both liver cancer and CNS
cancer. The infant incidence rate for
hepatoblastoma was 95% higher in whites
than in blacks (11.1 vs. 5.7 per million,
respectively) and for CNS malignancies the
rate was 120% higher for whites (37.7 per
million) than for blacks (17.1 per million).
Of the more common infant cancers, only
rates of malignant germ cell tumors and
retinoblastoma were higher in black in-
fants, and these differences were slight.
The small number of cases for black infants
precluded reliable subgroup comparisons
for the CNS cancers and leukemias.
Distribution by month of age at diagnosis
Although we lack a valid denominator
(the number of children at risk by month of
Figure XII.5: Total childhood cancer age-specific
incidence rates by race, both sexes
SEER, 1976-84 and 1986-94 combined
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Age (in years) at diagnosis
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
Average annual rate per million
White
Black
All races
+
)
%
Figure XII.6: Ratios of white to black incidence rates
among infants by type, both sexes
SEER, 1976-84 and 1986-94 combined
1.78
1.66
2.2
0.89
1.07
0.93
1.03
1.95
Neuroblastoma
Leukemias
CNS
Retinoblastoma
Wilms'
Germ cell
Soft tissue
Hepatic
0 0.5 1 1.5 2 2.5
Ratio of white to black incidence rates
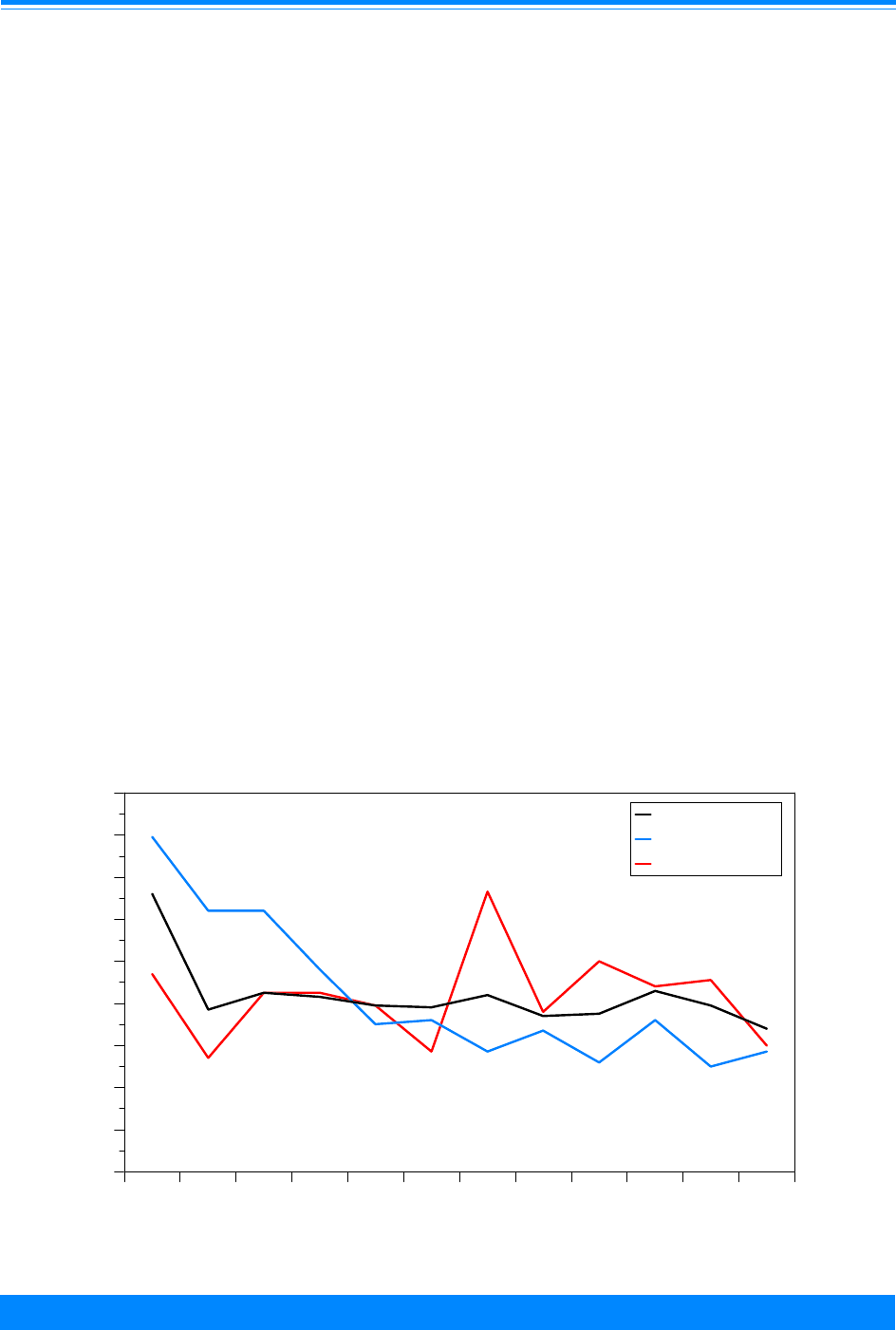
XII
154
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG INFANTS
age) to accurately calculate month-specific
age incidence rates, the percentage of
cancer cases (all types combined) by month
of age at diagnosis is presented in Figure
XII.7. This distribution shows that 13% of
the infant malignancies were diagnosed
during the first month following birth.
Separate distributions for all leukemias
combined and for neuroblastoma are also
illustrated. Approximately half of neuro-
blastomas were diagnosed within the first 4
months of life, with 16% diagnosed during
the month following birth. In fact, many of
these tumors were likely detected in utero.
Unlike neuroblastoma, the pattern of
diagnosis for leukemias shows that the
peak month of diagnosis occurred during
the latter part of infancy, in the 7
th
month
of life. Although not shown in the figure,
the majority of infant germ cell tumors
(56%) were diagnosed very soon after birth,
before 2 months of age. There was no other
cancer type that presented with such a
large percentage of cases so early after
birth.
TRENDS
As discussed in a previous report of
trends in infant cancer [2], considerable
caution must be exercised when interpret-
ing temporal changes in rates for a single
age group. Any trend analysis of incidence
rates may be confounded by changes in
population characteristics, the accuracy of
census estimates, screening practices,
diagnostic technology, morphology classifi-
cations, and case ascertainment. One or
more of these factors could effectively
conspire to show increasing incidence over
time that is not reflective of more cancer,
but rather of earlier diagnosis or better
case identification. Neuroblastoma is an
excellent example of this because of the
recent introduction of (controversial)
screening practices for earlier detection in
Japan and Canada [3,4] and because of the
Figure XII.7: Percent distribution of infant cancer cases
by month of age at diagnosis and type, all races
both sexes, SEER, 1976-84 and 1986-94 combined
,
,
,
,,
,
,
,
,
,
,
,
&
&
&&
&
&
&
&
&
&
&
&
#
#
#
#
#
#
#
#
#
#
#
#
0123456789101112
Age (in months) at diagnosis
0
2
4
6
8
10
12
14
16
18
Relative percent
All sites
Neuroblastoma
Leukemias
#
&
,

XII
155
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG INFANTS
fairly recent advent of fetal ultrasound
diagnosis [5]. With that caveat in place, a
table of incidence rates for two time periods
(1976-84 and 1986-94) is presented. (Table
XII.1) The percentage change in the inci-
dence rates is also shown.
These data imply that incidence rates
of infant cancer were higher during the
time period 1986-94 than the time period
1976-84. To what degree these data repre-
sent a true increase in cancer incidence in
the US, compared with the influence of the
potentially misleading factors that were
mentioned above, has not been determined
and requires more detailed assessment.
For instance, the 125% increase in germ cell
tumors is largely concentrated among non-
gonadal malignancies (primarily sacrococ-
cygeal and pelvic) among female infants
(see the monograph chapter entitled:
“Germ Cell, Trophoblastic and Other Go-
nadal Tumors” for additional details).
While this could indicate a true increase in
the incidence of this disease, it is likely that
a substantial proportion of the increase
reflects changes over time in the identifica-
tion by pathologists of malignant elements
in teratomas with an otherwise benign
appearance, leading to increased reporting
of malignant teratomas among infants [6].
Likewise, the increase in reported rates of
infant neuroblastoma, a disease with an
exceptionally high survival rate and with a
propensity for spontaneous regression, may
reflect increased diagnoses and reporting of
tumors that previously regressed before
being detected [7].
SURVIVAL
The prognosis for infants with cancer is
often worse than in children of older ages,
even when comparing the same histologic
diagnosis. For instance, the 5-year relative
survival for children younger than 15 years
of age who were diagnosed with acute
lymphoid leukemia from 1975-94 was well
over 70%, but for infants the survival rate
was 35%. Survival was bleak at all ages for
acute non-lymphoid leukemia, but it was
the poorest for infants, with a relative 5-
year survival of 30%. This pattern was also
evident for rhabdomyosarcomas and CNS
tumors, particularly ependymomas and
PNET. For ependymomas, 5-year relative
survival for infants was less than 20% and
for PNET it was less than 30%. Infant
neuroblastoma was an exception. Over
80% of children diagnosed with neuroblas-
toma during infancy were alive 5 years
following diagnosis. In contrast, for chil-
dren diagnosed with neuroblastoma at age
1 year or older, the 5-year relative survival
was about 45%. The 5-year survival from
1975-94 was also very good for children
diagnosed with Wilms’ tumor (86%) and
retinoblastoma (over 90%).
SUMMARY
The age of peak incidence of cancer in
children occurs during the first year of life.
Neuroblastoma is the most common infant
malignancy, followed by the leukemias and
the CNS cancers. Female infants and male
infants have essentially the same overall
cancer incidence rates. White infants have
substantially higher cancer rates than
black infants for most cancer types. Inci-
Table XII.1: Average annual incidence rates
per million by type, age <1, all races
both sexes, SEER, 1976-84 and 1986-94
Cancer type 1976-84 1986-94 % Change
All cancer 197.9 269.3 36
Neuroblastoma 55.2 74.4 35
All leukemia 35.9 45.4 26
All CNS 23.3 36.5 57
Retinoblastoma 22.1 31.5 43
Wilms’ 21.4 23.6 10
Germ cell 9.5 21.3 124
Soft tissue 13.6 16.6 22
Hepatic 7.6 11.4 50
Lymphoma 4.5 4.2 - 7
Epithelial 2.6 3.0 15
Other/Unspecified 1.4 1.0 -29
Bone 0.5 0.5 0

XII
156
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG INFANTS
dence rates were notably higher for the
period 1986-94 compared with 1976-84,
although many factors other than a real
increase in incidence may be influencing
this trend. Relative survival for infants is
very good for neuroblastoma, Wilms’ tumor
and retinoblastoma, but not for most other
types of cancer.
The distribution of malignant disease
in infants is quite different from that which
is found in older children, adolescents, or
adults. For instance, embryonal tumors
such as neuroblastoma, Wilms’ tumor,
retinoblastoma, medulloblastoma and
hepatoblastoma are more prevalent in
infants than in humans of any other age.
The descriptive epidemiologic data that is
presented here may serve to stimulate
ideas for further etiologic research into the
multifactorial nature of cancer occurrence.
For example, the initial two-hit theory for
carcinogenesis was developed primarily
from clinical observations of a higher
frequency of bilaterality of retinoblastoma
in infants than in older children [8,9]. The
study of infant cancer can aid in developing
new hypotheses related to how aberrant
genetic processes, early developmental
abnormalities and gene-environment
interactions contribute to the carcinogenic
process. The study of retinoblastoma, and
later of Wilms’ tumor, led to the discovery of
two important tumor suppressor genes that
are related to adult as well as pediatric
malignancy [10]. Recent work has shown
that hematologic malignancies manifest
differently in infants than in older children
[11]. All these factors speak to the impor-
tance of further research into the epidemi-
ology and biology of cancer in very young
children.
Reference List
1. Kenney LB, Reaman GH; Special consider-
ations for the infant with cancer. Pizzo P,
Poplack D, Editors. Principles and Practices of
Pediatric Oncology. 3rd ed. Philadelphia, PA:
Lippincott-Raven; 1997:343-356.
2. Gurney JG, Ross JA, Wall DA, Bleyer WA,
Severson RK, Robison LL. Infant cancer in the
US: histology-specific incidence and trends,
1973 to 1992. J Pediat Hematol Oncol.
1997;19:428-432.
3. Bessho F. Effects of mass screening on age-
specific incidence of neuroblastoma. Int J
Cancer. 1996;67:520-522.
4. Woods WG, Tuchman M, Robison LL, et al.
Screening for neuroblastoma is ineffective in
reducing the incidence of infavourable ad-
vanced stage disease in older children. Eur J
Cancer. 1997;33:2106-2112.
5. Holgersen LO, Subramanian S, Kirpekar M,
Mootabar H, Marcus JR. Spontaneous resolu-
tion of antenatally diagnosed adrenal masses.
J Pediatr Surgery. 1996;31:153-155.
6. Hawkins, e, Issacs, H. Et al. (1993). “Occult
malignancy in neonatal sacrococcygeal terato-
mas. A report from a Combined Pediatric
Oncology Group and Children’s Cancer Group
study”. Am J Pediatr Hematol Oncol 15(4);406-
9.
7. Brodeur GM, Castleberry RP. Neuroblastoma.
Pizzo P, Poplack D, Editors. Principles and
Practices of Pediatric Oncology. 3rd ed. Phila-
delphia, PA: Lippincott-Raven; 1997:761-.797.
8. Knudson AG. Mutation and cancer: statistical
study of retinoblastoma. Proc Natl Acad Sci
USA. 1971;68:820.
9. Pollock BH, Knudson AG; Preventing cancer in
adulthood: advise for the pediatrician. Pizzo P,
Poplack D, Editors. Principles and Practices of
Pediatric Oncology. 3rd ed. Philadelphia, PA:
Lippincott-Raven; 1997:1421-1435.
10. Malkin D, Portwine C. The genetics of child-
hood cancer. Eur J Cancer. 1994;30A:1942-
1946.
11. Ross JA, Davies SM, Potter JD, Robison LL.
Epidemiology of childhood leukemia, with a
focus on infants. Epidemiol Rev. 1994;16:243-
272.

XIII
157
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
HIGHLIGHTS
Incidence
♦ The incidence of cancer among adolescents (i.e., 15-19 year-olds) in SEER areas for
1986-95 was 202.2 per million, which was similar to the incidence of cancer among
0-4 year-olds and substantially greater than the incidence for 5-9 and 10-14 year-
olds (Table XIII.1).
♦ The spectrum of cancers that occurred among 15-19 year-olds was distinctive from
those that occurred in young children. For SEER areas from 1986-95, the most
common tumors among adolescents were Hodgkin’s disease (16.1%), germ cell
tumors (15.2%), CNS tumors (10.0%), non-Hodgkin’s lymphoma (NHL) (7.6%),
thyroid cancer (7.2%), malignant melanoma (7.0%), and acute lymphoblastic leuke-
mia (ALL) (6.4%) (Figure XIII.1 and Table XIII.1).
♦ The embryonal cancers that predominated among young children (e.g., neuroblas-
toma, Wilms tumor, retinoblastoma, and hepatoblastoma) (Figure XIII.2) were
distinctly uncommon among 15-19 year olds (Figure XIII.1 and Table XIII.1).
Trends in Incidence
♦ The annual incidence of cancer for adolescents increased from 183.0 per million in
1975-79 and to 203.8 per million in 1990-95 (Table XIII.4 and Figure XIII.3).
♦ The largest contributor to this increase was the germ cell, trophoblastic, and other
gonadal tumor category (specifically testicular and ovarian germ cell tumors).
♦ Smaller increases in incidence were observed for non-Hodgkin’s lymphoma (NHL),
osteosarcoma, and acute lymphoblastic leukemia (ALL).
♦ No significant increases in incidence were observed for CNS tumors, melanoma,
thyroid cancer, Hodgkin’s disease, or soft tissue sarcomas.
Incidence by Gender and Race
♦ Rates of specific cancer types differed substantially by gender and by race among
adolescents.
♦ For gender, these differences were most remarkable for thyroid cancer (much more
common in females) and for the bone tumors, ALL, and NHL (the latter three more
common among males) (Table XIII.2).
♦ Black 15-19 year olds had much lower incidence rates for Ewing’s sarcoma, testicu-
lar germ cell tumors, and melanoma than did whites. Black adolescents also had
modestly lower incidence of ALL and thyroid cancer compared to white 15-19 year
olds (Table XIII.3).
Survival
♦ Overall 5-year survival rates for adolescents with cancer improved from 69% to 77%
from 1975-84 to 1985-94 (Table XIII.5).
♦ For some cancer types (Hodgkin’s disease, germ cell tumors, thyroid cancer, and
melanoma), 5-year survival rates were 90% or better for the most recent time period
(1985-94).
♦ For other cancer types (e.g., osteosarcoma, Ewing’s sarcoma, ALL, and AML)
survival rates for adolescents remained less than 60%.
Malcolm A. Smith, James G. Gurney, Lynn A. Gloeckler Ries

XIII
158
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
Table XIII.1: Age-specific cancer incidence rates per million and percentage of total cases by ICCC
category and age group, all races, both sexes, SEER, 1986-95
Age (in years) at diagnosis
Tumor category
<5
Rate
5-9
Rate
10-14
Rate
15-19
Rate
% of Total for
15-19 Group
All Sites 199.9 110.2 117.3 202.2 100.0%
Acute lymphoblastic leukemia (ALL) 58.2 30.3 17.8 12.9 6.4%
Acute myeloid leukemia (AML) (Ib) 10.1 4.5 5.7 8.5 4.2%
Hodgkin's disease (IIa) 0.8 3.9 11.7 32.5 16.1%
Non-Hodgkins lymphoma (NHL) (IIb,c,e) 5.9 8.9 10.3 15.3 7.6%
CNS tumors (III(total)) 36.0 31.9 24.6 20.2 10.0%
Ependymoma (IIIa) 5.6 1.6 1.3 1.1 0.5%
Astrocytoma (IIIb) 15.0 15.9 15.1 12.3 6.1%
Medulloblastoma/PNET (IIIc) 9.6 7.3 4.0 2.5 1.2%
Neuroblastoma & ganglioneuroblastoma (IVa) 27.4 2.6 0.8 0.5 0.2%
Retinoblastoma (V(total)) 12.5 0.5 0.0 0.1 0.0%
Wilms', rhabdoid, clear cell sarcoma (VIa) 18.0 5.8 0.6 0.4 0.2%
Hepatic tumors (VII(total)) 4.8 0.4 0.4 1.0 0.5%
Hepatoblastoma (VIIa) 4.6 0.2 0.1 0.0 0.0%
Osteosarcoma (VIIIa) 0.3 2.8 8.3 9.4 4.6%
Ewing's sarcoma (VIIIc) 0.3 1.9 4.1 4.6 2.3%
Soft tissue sarcoma (IX(total)) 10.9 8.3 10.9 15.9 7.9%
Rhabdomyosarcoma and embryonal sarcoma (IXa) 6.5 4.4 3.5 3.9 1.9%
Non-rhabdo soft tissue sarcoma (IXb-e) 4.4 4.0 7.4 11.9 5.9%
Germ Cell, trophoblastic, & other gonadal tumors (X (total)) 6.9 2.4 6.7 30.8 15.2%
Thyroid carcinoma (XIb) 0.1 1.0 4.1 14.6 7.2%
Malignant melanoma (XId) 0.8 0.6 2.8 14.1 7.0%
Other and unspecified carcinomas (XIf) 0.4 0.8 2.8 10.5 5.2%
INTRODUCTION
The adolescent population (here de-
fined as age 15-19 years) have variably
been included in analyses and reports of
childhood cancer. An NIH Policy concerning
inclusion of children in clinical research
defines children as being younger than 21
years of age, while the Food and Drug
Administration considers children to be 15
years and younger. Regardless of the
definition of children that is applied for
regulatory or reporting purposes, it is
instructive to consider the 15-19 year old
population separately because the types of
tumors that occur in this population differ
substantially from those that predominate
in younger children and in adults. Addi-
tionally, the 15-19 year old age group is one
whose participation rate in cancer clinical
trials has been noted to be much lower
than that for younger children [1].
In this chapter, differences in cancer
types and their incidence between the 15-
19 year group and younger children will be
highlighted. Additional points for emphasis
are the changes in cancer incidence for this
older age group from 1975 to 1995 and the
distinctive sex distribution for individual
tumor types. The chapter concludes with a
summary of survival rates for the 15-19
year old population, illustrating that
survival for many tumor types has im-
proved from 1975-84 to 1985-94.
INCIDENCE
Distribution of tumor types by 5-year age
groups
The incidence of specific cancers by
International Classification of Childhood
Cancer (ICCC) codes for the period 1986-
1995 is shown in Table XIII.1 by 5-year age
groups. Figure XIII.1 shows that the most
common tumors in the adolescent popula-
tion were Hodgkin’s disease (16.1%), germ
cell tumors (15.2%), CNS tumors (10.0%),

XIII
159
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
Figure XIII.1: Distribution of cancer types, age 15-19
all races, both sexes, SEER, 1986-95
Hodgkin's
16.1%
NHL
7.6%
Germ cell
15.2%
CNS
10.0%
Thyroid
7.2%
Melanoma
7.0%
ALL
6.4%
AML
4.2%
Non-RMS sarcoma
5.9%
Osteosarcoma
4.6%
Ewing's
2.3%
Rhabdomyosarcoma (RMS)
1.9%
Other
11.6%
non-Hodgkin’s lymphoma (NHL) (7.6%),
thyroid cancer (7.2%), malignant melanoma
(7.0%), and acute lymphoblastic leukemia
(ALL) (6.4%). Table XIII.1, as well as
comparison of Figures XIII.1 and XIII.2,
illustrates that a group of tumors that
occurred commonly among children
younger than 5 years of age were virtually
absent among 15-19 year olds, including:
neuroblastoma, Wilms’ tumor, retinoblas-
toma, ependymoma, and hepatoblastoma.
These 5 tumor types accounted for approxi-
mately 35% of cases among children
younger than 5 years of age (Figure XIII.2),
Figure XIII.2: Distribution of cancer types, age <5
all races, both sexes, SEER, 1986-95
ALL
29.1%
AML
2.7%
CNS
18.0%
Neuroblastoma
13.7%
Wilms'
9.0%
Retinoblastoma
6.3%
Other
7.0%
Germ cell
3.4%
Rhabdomyosarcoma (RMS)
3.3%
NHL
3.0%
Hepatoblastoma
2.3%
non-RMS sarcoma
2.2%

XIII
160
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
but less than 1% of cases among 15-19 year
olds.
The distribution of tumor types within
several ICCC categories for 15-19 year olds
compared with children younger than 15
years of age warrants specific comment.
The distribution of soft tissue sarcoma
diagnoses differed by age. Rhabdomyosar-
coma accounted for 60% of soft tissue
sarcoma cases among children younger
than 5 years of age. However, the incidence
of rhabdomyosarcoma decreased with age,
while the incidence of non-rhabdomyosar-
coma soft tissue sarcomas increased with
age, so that among 15-19 year olds, rhab-
domyosarcoma accounted for only 25% of
soft tissue sarcoma diagnoses (Table
XIII.1). The annual incidence of ALL also
decreased with age: among children
younger than 5 years of age the rate was
58.2 per million, while for 15-19 year olds
the rate was nearly 5-fold less (12.9 per
million) (Table XIII.1). Although ALL
accounted for nearly 30% of cancer cases
among children younger than 5 years of
age, it represented only 6.4% of cases
among the 15-19 year olds. The incidence
of NHL was higher among 15-19 year olds
than among younger age groups (Table
XIII.1). This increase was largely the
result of much higher rates for diffuse large
cell lymphoma among 15-19 year olds,
while rates for Burkitt’s lymphoma and
lymphoblastic lymphoma for 15-19 year
olds were similar to or less than those
observed for children less than 15 years of
age (see Lymphoma chapter for details).
Sex-specific incidence
The overall incidence of cancer cases
was similar among males and females in
the 15-19 year old age group for the years
1986 to 1995 (Table XIII.2). However, the
overall similarity masked marked differ-
ences in rates for individual tumor types.
There was a strong male predominance for
ALL, NHL, osteosarcoma, and Ewing’s
sarcoma, with 60% to over 100% higher
rates occurring in males than females.
Additionally, rates of CNS tumors and germ
cell tumors were 30-40% higher in males
than in females. On the other hand, there
was a female predominance for Hodgkin’s
disease, thyroid carcinoma, and melanoma.
The rates by sex for the 15-19 year olds
were distinct from those for children
younger than 15 years of age. For ALL,
there was only a 20% excess of male cases
among the children younger than 15 years
of age, compared to a nearly 120% excess
for the 15-19 year olds. There was a male
predominance for Hodgkin’s disease in the
0-14 year olds, compared to a female
predominance in 15-19 year olds. The
situation was reversed for the germ cell,
trophoblastic, and other gonadal tumors,
which were more common among females
Table XIII.2: Age-adjusted cancer incidence rates per million by ICCC group
sex, and age, all races, both sexes, SEER, 1986-95
Age (in years) at diagnosis
Tumor category
0-14
Male*
0-14
Female*
0-14
Ratio: M/F
15-19
Male
15-19
Female
15-19
Ratio: M/F
ALL SITES 149.5 128.7 1.2 204.3 199.9 1.0
Acute lymphoblastic leukemia 37.1 30.9 1.2 17.5 8.0 2.2
Acute myeloid leukemia (Ib) 6.6 6.5 1.0 8.4 8.5 1.0
Hodgkin's disease (IIa) 6.5 5.0 1.3 28.8 36.5 0.8
Non-Hodgkin's lymphoma (IIb,c,e) 12.3 4.5 2.7 19.4 11.0 1.8
CNS (III) 33.0 27.9 1.2 23.0 17.3 1.3
Osteosarcoma (VIIIa) 3.8 4.3 0.9 11.5 7.1 1.6
Ewing's sarcoma (VIIIc) 2.3 2.2 1.1 5.8 3.3 1.8
Soft tissue sarcomas (IX) 10.9 9.1 1.2 17.4 14.3 1.2
Germ cell tumors (X) 4.3 6.2 0.7 35.2 26.1 1.4
Thyroid carcinoma (XIb) 0.9 2.9 0.3 3.7 26.2 0.1
Melanoma (XId) 1.3 1.6 0.8 10.5 17.9 0.6
*Adjusted to the 1970 US standard population

XIII
161
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
in children younger than 15 years of age,
but more common among males in the 15-
19 year old age group. The male predomi-
nance for bone tumors observed in adoles-
cents was absent in the younger than 15
year old age group. For NHL, there was a
marked male predominance for both 0-14
year olds and for 15-19 year olds.
Black-white differences in incidence
The incidence of cancer among whites
age 15-19 years for 1986-95 was
approximately 1.5-fold higher than that
among blacks age 15-19 years (Table
XIII.3). In comparing cancer incidence for
white and black 15-19 year olds, incidence
rates at least 2-fold higher were observed
among whites, compared to blacks for ALL,
germ cell tumors, thyroid cancer, Ewing’s
sarcoma, and melanoma. The low incidence
for germ cell tumors among blacks was
restricted to testicular germ cell tumors in
males. White females and black females
had similar rates for germ cell tumors (see
Germ Cell, Trophoblastic, and Other
Gonadal Tumor chapter for additional
details). While the very high ratio of white
to black cases for melanoma may be
explained by the protection afforded from
ultraviolet light by melanin, the reasons
that blacks have lower rates of Ewing’s
sarcoma, ALL, testicular germ cell tumors,
and thyroid cancer are not apparent.
TRENDS
The average annual age-adjusted
cancer incidence among 15-19 year olds
increased from 183 per million in 1975-79
to slightly over 203.8 per million in 1990-95
(Figure XIII.3 and Table XIII.4). By
comparison, the incidence of cancer for
children younger than 15 years of age
increased from 124.3 per million in 1975-79
to 139.9 per million in 1990-95. The
greatest numeric increase in annual
incidence for the 15-19 year group occurred
for the germ cell,trophoblastic, and other
gonadal (GCTOG) tumors. This increase
was primarily the result of an increase in
the incidence of testicular germ cell tumors
among males (increasing from 22.1 to 28.4
per million) and ovarian germ cell tumors
among females (increasing from 7.9 to 13.3
Table XIII.3: Age-specific cancer incidence rates
per million by ICCC group and race
age 15-19, SEER, 1986-95
Tumor category White Black
W/B
Ratio
Total 213.5 144.8 1.5
Acute lymphoblastic leukemia 14.3 6.4 2.2
Acute myeloid leukemia (Ib) 8.3 7.1 1.2
Hodgkin's disease (IIa) 36.5 26.9 1.4
Non-Hodgkin's lymphoma (IIb,c,e) 16.1 9.4 1.7
CNS (III) 21.8 15.8 1.4
Osteosarcoma (VIIIa) 9.2 8.4 1.1
Ewing's sarcoma (VIIIc) 5.4 0.3 18.0
Soft tissue sarcomas (IX) 14.5 20.5 0.7
Germ cell tumors (X) 33.9 13.8 2.5
Thyroid carcinoma (XIb) 15.5 6.7 2.3
Melanoma (XId) 16.1 0.3 53.7
Figure XIII.3: Trends in age-adjusted* cancer incidence
rates by age group, all races, both sexes, SEER, 1975-95
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
(
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
75 80 85 90 95
Year of diagnosis
0
50
100
150
200
Average annual rate per million
Incidence 15-19
Incidence <15
&
(
*Adjusted to the 1970 US standard population

XIII
162
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
per million). As discussed in the chapter for
GCTOG tumors, the increase in gonadal
carcinomas is likely artifactual and
attributable to changes in reporting of
ovarian tumors during this time period,
specifically inclusion of borderline tumors of
the ovary. The incidence of ALL, NHL, and
osteosarcoma also increased from 1975-79
to 1990-95 (Table XIII.4). These four tumor
types accounted for the majority of the
increase in cancer incidence for the 15-19
year old group. No significant increases or
decreases in incidence were observed for
CNS tumors, melanoma, thyroid cancer,
Hodgkin’s disease, or soft tissue sarcomas.
SURVIVAL
Table XIII.5 shows 5-year relative
survival rates for different cancer types for
15-19 year olds , with comparison made
between an earlier time period (1975-84)
Table XIII.4: Average annual age-specific incidence rates per million adolescents
15-19 years old for selected tumors, all races, both sexes, SEER, 1975-95
Tumor type (ICCC Category) 1975-79
Rate
1980-84
Rate
1985-89
Rate
1990-95
Rate
All Sites 183.0 187.7 199.3 203.8
Acute Lymphoblastic Leukemia (Ia) 10.6 13.2 12.4 13.0
Non-Hodgkin’s lymphoma (IIb,c,e) 10.7 14.5 14.4 16.3
Osteosarcoma (VIIIa) 6.6 8.9 9.7 9.3
Germ cell, trophoblastic and other gonadal tumors (X) 23.2 24.0 28.6 32.0
Testicular germ cell tumor (Xc, male) 22.1 26.7 24.9 28.4
Ovarian germ cell tumor (Xc, female) 7.9 8.3 11.8 13.3
Gonadal carcinoma (Xd) 2.7 2.4 4.3 5.3
Rates are per 1,000,000.
Table XIII.5: 5-Year Relative Survival Rates by ICCC group
and time period, age 15-19, all races, both sexes
SEER, 1975-84 and 1985-94
TUMOR CATEGORY 1975-84 1985-94
Total 69% 77%
Acute lymphoblastic leukemia (ALL) 35% 51%
Acute myeloid leukemia (AML) (Ib) 22% 42%
Hodgkin's (IIa) 88% 90%
NHL (IIb,c,e) 56% 69%
Astrocytoma (IIIb) 62% 75%
Medulloblastoma (IIIc) 63% 75%
Osteosarcoma (VIIIa) 49% 59%
Ewing's sarcoma (VIIIc) 36% 56%
Soft tissue sarcoma (IX) 70% 63%
Rhabdomyosarcoma (IXa) 40% 45%
Germ cell tumors (X) 79% 90%
Thyroid carcinoma (XIb) 99% 99%
Melanoma (XId) 84% 92%

XIII
163
National Cancer Institute SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
and a recent reporting period (1985-94).
Important observations concerning survival
rates include:
• For all cancer diagnoses in the 15-
19 year old age group, the 5-year
survival rate for the recent
reporting period was 77%, which
was higher than that for the other
five-year age groups younger than
20 years of age.
• Five-year survival rates of 90% or
higher were observed for Hodgkin’s
disease, germ cell tumors, thyroid
carcinoma, and melanoma. For
germ cell tumors and melanoma,
the survival rates improved
between the earlier and recent
time period.
• Five-year survival rates for NHL
improved between 1975-84 and
1985-94 from approximately 56%
to 69%.
• Five-year survival rates for both
ALL and AML improved substan-
tially for the 15-19 year old group,
with survival rates for 1985-94 of
51% and 42%, respectively, com-
pared to only 35% and 22% for
1975-84.
Five-year survival rates for Ewing’s
sarcoma improved from 36% to 56%
between the earlier and the recent report-
ing period.
The mortality burden is a function of
the survival and the incidence rates. The
leukemias are the primary contributor to
the cancer mortality burden for cancers
developing in the 15-19 year olds. In
addition to leukemia, bone cancer, soft
tissue sarcoma, CNS cancer, NHL, and
Hodgkin’s disease are the most common
causes of cancer death among this group -
see the mortality chapter. Although thyroid
carcinoma and melanoma are among the
more common cancers in this age group,
they contribute little to the overall cancer
mortality burden for the 15-19 year old age
group.
SUMMARY
The spectrum of malignancies that
occur in adolescents is distinctive when
compared to those that occur in young
children and those that occur in older
adults. The embryonal cancers that
predominate among young children (e.g.,
neuroblastoma, Wilms’ tumor, retinoblas-
toma, ependymoma, and hepatoblastoma)
are very uncommon among 15-19 year olds.
Similarly, the epithelial carcinomas of
adults (e.g., lung, breast, colon) rarely occur
in 15-19 year olds. While some types of
acute leukemias and CNS cancers are
shared with both the older adult and the
young childhood populations, the 15-19
year old group experiences high rates of a
set of tumors (including germ cell tumors,
Hodgkin’s disease, and the bone cancers)
that are relatively characteristic of the
adolescent/young adult age group.
The annual incidence of cancer in
adolescents increased from 183 per million
for 1975-79 to 203.8 per million from 1990-
95. The largest contributor to this increase
was the germ cell, trophoblastic, and other
gonadal tumor category (specifically
testicular and ovarian germ cell tumors),
with smaller contributions from NHL,
osteosarcoma, and ALL.
Rates of specific cancer types differed
substantially by sex and by race. For sex,
these differences were most remarkable for
thyroid cancer (much more common in
females) and for the bone tumors, ALL, and
NHL (the latter three more common among
males). Black 15-19 year olds had much
lower incidence rates of Ewing’s sarcoma,
testicular germ cell tumors, and melanoma
than did whites, and modestly lower

XIII
164
National Cancer Institute
SEER Pediatric Monograph
CANCER AMONG ADOLESCENTS 15-19 YEARS OLD
incidence rates of ALL and thyroid cancer.
Five-year survival for 15-19 year olds
increased from 69% to 77% from 1975-84 to
1985-94, with a 90% survival rate or better
for several diagnoses (Hodgkin’s disease,
germ cell tumors, thyroid cancer, and
melanoma). However, for some cancers, the
survival rates remained less than 60%
(including osteosarcoma, Ewing’s sarcoma,
ALL, and AML).
Reference List
1. Bleyer W, Tejada H, Murphy S, et al: National
cancer clinical trials: Children have equal
access; adolescents do not. J Adolesc Health
21:366-373, 1997.

XIV
165
National Cancer Institute SEER Pediatric Monograph
CHILDHOOD CANCER MORTALITY
Introduction
Over the past two decades, childhood
cancer mortality in the United States has
declined dramatically. To present a compre-
hensive picture of childhood cancer occur-
rence and outcome, it would be ideal to
include cancer-specific data on incidence,
survival, and mortality within each indi-
vidual chapter of the monograph. The
available data on mortality, however, are
obtained from death certificates and col-
lected by the National Center for Health
Statistics (NCHS) for the entire United
States. In addition to the difference in
geographic coverage between NCHS and
SEER areas, the cancer classification used
by NCHS for mortality is less specific than
that used by SEER areas. Therefore, we
are presenting this separate chapter on
cancer mortality and have included inci-
dence [1,2] comparisons based on compa-
rable definitions to the mortality data [3].
A further explanation on differences be-
tween the incidence definitions used in the
other chapters and mortality is included at
the end of this chapter. The mortality data
are provided by the National Center for
Health Statistics to the National Cancer
Institute on public-use tapes.
All Sites
In contrast to incidence rates, cancer
mortality declined substantially between
1975 and 1995 (Figure XIV.1). There were
statistically significant declines in
mortality for each of the five-year age
groups ( <5, 5-9, 10-14, and 15-19) for
cancers combined. The declines by age
group ranged from 2.0 to 3.2 percent per
year. The overall decline in mortality was
nearly 40 percent between 1975 and 1995,
a statistically significant decrease of 2.6
percent per year. The overall incidence
increased 0.8 percent per year. There were
2,275 cancer deaths among children in
1995. Except for those 15-19, leukemia and
brain/other nervous system comprised more
than 50 percent of the deaths due to cancer
(Figure XIV.2). The relative difference for
Lynn A. Gloeckler Ries
Figure XIV.1: Trends in childhood cancer age-adjusted*
rates, all races, both sexes, age <20
SEER incidence & US mortality, 1975-95
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&&
&
&
&
&
&
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
1975 1980 1985 1990 1995
Year of diagnosis/death
0
25
50
75
100
125
150
175
Average annual rate per million
Incidence
Mortality
*Adjusted to the 1970 US standard population
Figure XIV.2: Percent distribution of childhood cancer
mortality by type and age group, age <20
all races, both sexes, United States, 1995
32
37
40
29
34
25
31
24
14
23
43
31
36
57
43
<5
5-9
10-14
15-19
<20
Age (in years) at diagnosis
0 20406080100
Percent
Leukemias Brain/ONS Other

XIV
166
National Cancer Institute
SEER Pediatric Monograph
CHILDHOOD CANCER MORTALITY
the 15-19 year olds was due to deaths from
lymphoma (14%), bone (13%), and soft
tissue sarcomas (9%). Leukemias and
brain cancer, however, accounted for 57% of
cancer deaths for all children combined.
Leukemia
In 1995, thirty-four percent of the
childhood cancer deaths were due to leuke-
mia. The death rate from leukemia fell
nearly 50 percent between 1975 and 1995
(Figure XIV.3), a statistically significant
decline of 3.4 percent per year while the
incidence increased between 1975 and
1995. Mortality rates declined significantly
for each of the age groups (<5, 5-9,10-14,
15-19, <20) and for both males and females.
Brain/other central nervous system (brain/
CNS)
In 1995, nearly one-fourth of childhood
cancer deaths were due to invasive malig-
nancies of the central nervous system,
primarily the brain. Mortality from brain
and other CNS cancer declined an average
of 1.1 percent per year. This was an overall
decline of 23 percent between 1975 and
1995 (Figure XIV.4). This mortality decline
occurred while the incidence rate increased
mainly in the mid-1980s [4].
Unlike most benign tumors,
noninvasive tumors of the brain/CNS have
the potential to be fatal. Figure XIV.5
illustrates mortality rates for brain tumors
classified as invasive, unspecified or uncer-
tain, and benign. If the behavior of the
Figure XIV.3: Trends in childhood leukemia age-adjusted*
rates, age <20, all races, both sexes
SEER incidence, and US mortaility, 1975-95
,
,
,
,
,
,
,
,,
,
,,
,
,
,
,
,
,
,
,
,
&
&
&
&
&
&
&
&&
&
&
&
&
&
&
&
&
&
&
&
&
1975 1980 1985 1990 1995
Year of diagnosis/death
0
10
20
30
40
50
Average annual rate per million
Incidence
Mortality
*Adjusted to the 1970 US standard population
Figure XIV.4: Trends in brain/other nervous system
cancer age-adjusted* rates, all races, both sexes
age <20, SEER incidence & US mortality, 1975-95
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,,
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
&
1975 1980 1985 1990 1995
Year of diagnosis/death
0
5
10
15
20
25
30
35
Average annual rate per million
Mortality
Incidence
*Adjusted to the 1970 US standard population
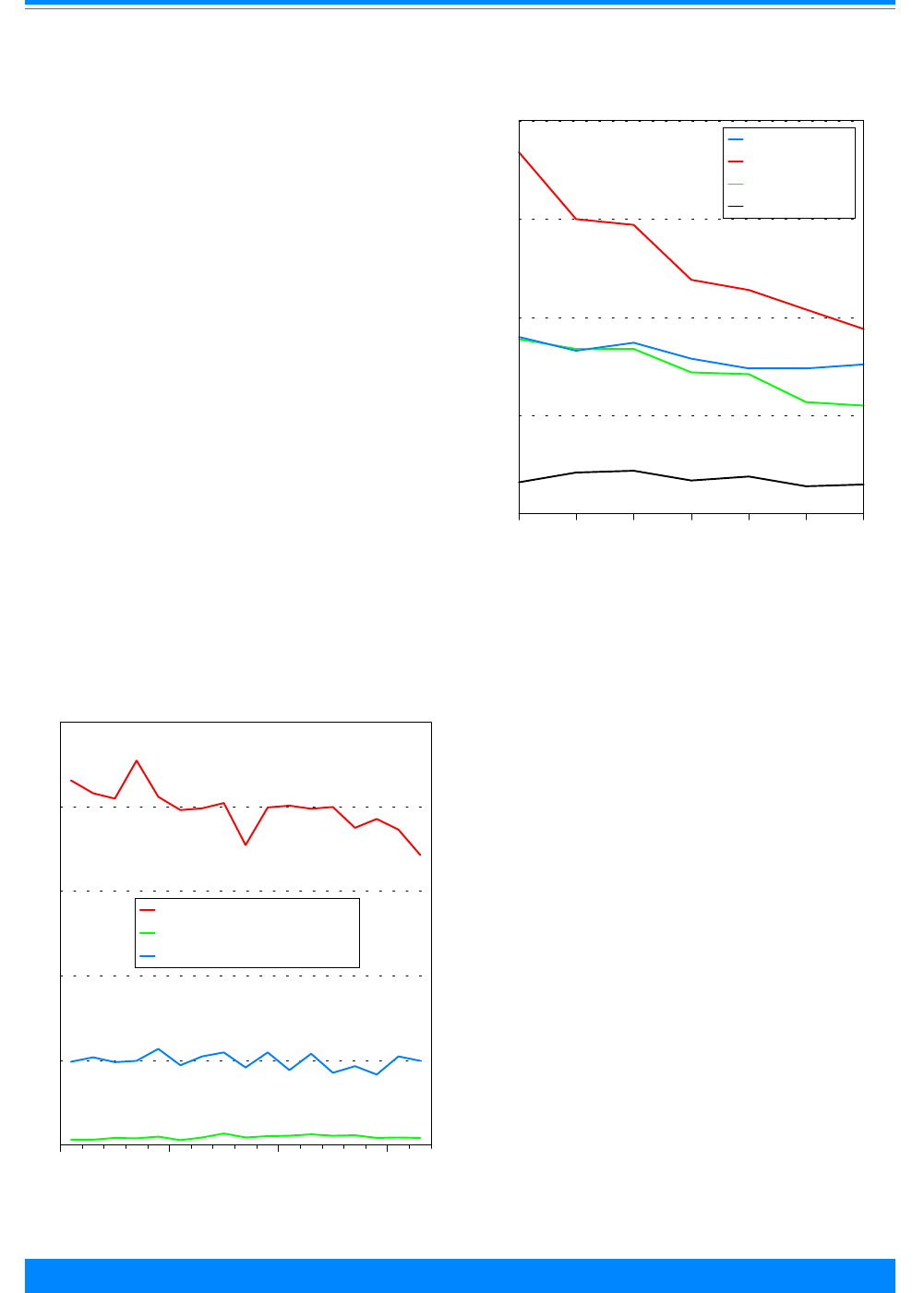
XIV
167
National Cancer Institute SEER Pediatric Monograph
CHILDHOOD CANCER MORTALITY
tumor is not clear from the death certifi-
cate, it is considered “unspecified or uncer-
tain”. Some of these tumors will be inva-
sive and some will not. Although mortality
from invasive tumors has declined some-
what over the past decade, there appears to
be no change over time in the rates of death
from brain tumors classified as either
“benign” or “unspecified or uncertain”.
Thus, the reduction in mortality from
invasive brain cancer does not appear to be
an artifact due to changes in the reporting
of the other categories of brain tumors. To
avoid changes in death classification be-
tween 1978 and 1979, this figure begins in
1979.
Ages 0-4
From 1975 to 1995, death rates from
cancer declined 2.9 percent per year among
children younger than 5 years of age. The
Figure XIV.6 shows the mortality rates for
the four leading causes of cancer death
among young children. The death rates
have declined for each. For leukemias, the
death rates declined by an average of 3.5
percent each year or more than 50 percent
between 1975 and 1995. After leukemia
and brain/CNS cancer, endocrine malignan-
cies were responsible for the most cancer
deaths. Most of the cancers classified as
“endocrine” in this age group were neuro-
blastomas. In 1995,there were 558 deaths
due to cancer among children younger than
5 years of age in the entire United States.
Ages 5-9
There were 523 deaths due to cancer
among children 5-9 years of age in the
entire United States in 1995. The age
group 5-9 years of age had the largest
decline in cancer mortality. The top four
mortality sites were leukemia, brain/CNS,
endocrine and non-Hodgkin’s lymphoma.
Figure XIV.6: Trends in age-specific cancer mortality
rates by type, age <5, all races, both sexes
United States, 1975-95
#
#
#
#
#
#
#
(
(
((
(
(
(
,
,
,
,
,
,
,
&
&
&
&
&
&&
1975-77 1981-83 1987-89 1993-95
Year of death
0
5
10
15
20
Average annual rate per million
Brain & ONS
Leukemia
Endocrine
Soft tissue
&
,
(
#
Figure XIV.5: Trends in age-adjusted* brain tumor
mortality rates by behavior, age <20, United States, 1979-95
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
)
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
%
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
$
1979 1984 1989 1994
Year of death
0
2
4
6
8
10
Average annual rate per million
Invasive
Benign
Unspecified & uncertain
$
%
)
*Adjusted to the 1970 US standard population
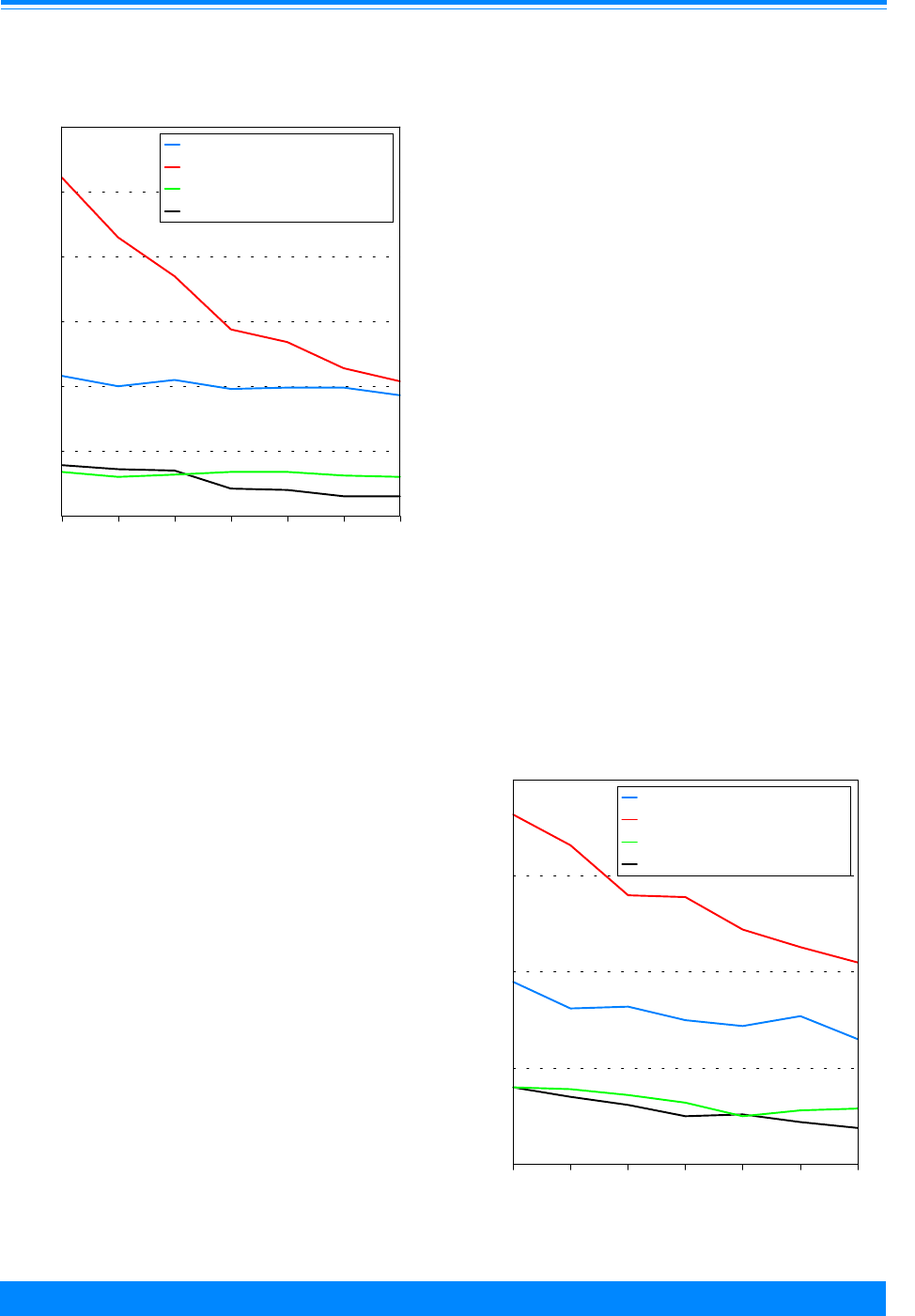
XIV
168
National Cancer Institute
SEER Pediatric Monograph
CHILDHOOD CANCER MORTALITY
The decline in leukemia deaths was 5
percent per year (Figure XIV.7).
Ages 10-14
There were 503 deaths due to cancer
among children 10-14 years of age in the
entire United States in 1995. The death
rate declined 2.5 percent per year. The
decline for leukemias was 3 percent per
year. The top four mortality sites were
leukemia, brain/CNS, bone/joints, and non-
Hodgkin’s lymphoma (Figure XIV.8).
Ages 15-19
There were 691 deaths due to cancer
among children 15-19 in the entire United
States in 1995. The overall cancer death
rate declined 2 percent per year. The top
five cancer mortality sites are shown for
this age group since the death rates for soft
tissue and non-Hodgkin’s lymphoma were
similar for the most recent time period
(Figure XIV.9).
Recent cancer mortality (1990-1995) by
race/ethnicity
The cancer mortality rates for all races
combined and for white children declined
2.4 and 3.0 percent per year, respectively.
The mortality rates for black and for His-
panic children declined 0.5 percent per year
between 1990 and 1995. For American
Indian children and Asian Pacific Islander
children, the death rates increased slightly
at 0.5 percent per year. The cancer death
rates for American Indian children (23.8
per million) and for Asian Pacific Islander
children (29.2 per million) were less than
those for white children (32.9), black chil-
dren (32.5) or Hispanic children (33.5 per
million children). The mortality data are
for the whole United States except for
Hispanics for which four states (New
Hampshire, Oklahoma, Connecticut and
Louisiana) are excluded. Hispanics can be
of any race and are therefore, not mutually
exclusive from the other categories.
Figure XIV.7: Trends in age-specific cancer mortality
rates by type, age 5-9, all races, both sexes
United States, 1975-95
#
#
#
#
#
#
#
(
((
(
((
(
,
,
,
,
,
,
,
&
&
&
&
&
&&
1975-77 1981-83 1987-89 1993-95
Year of death
0
5
10
15
20
25
30
Average annual rate per million
Brain & ONS
Leukemia
Endocrine
Non-Hodgkin's lymphomas
&
,
(
#
Figure XIV.8: Trends in age-specific cancer mortality
rates by type, age 10-14, all races
both sexes, United States, 1975-95
#
#
#
#
#
#
#
(
(
(
(
(
(
(
,
,
,
,
,
,
,
&
&
&
&
&
&
&
1975-77 1981-83 1987-89 1993-95
Year of death
0
5
10
15
20
Average annual rate per million
Brain & ONS
Leukemia
Bones & joints
Non-Hodgkin's lymphomas
&
,
(
#

XIV
169
National Cancer Institute SEER Pediatric Monograph
CHILDHOOD CANCER MORTALITY
Problems comparing incidence to mortality
The histology site groupings presented in
other chapters of this monograph are based
on the International Childhood Cancer
Classification (ICCC) [5]. While they are
useful groupings for incidence, there are
problems when comparing incidence to
mortality. The ICCC uses histology as its
main criteria and secondarily primary site.
The underlying cause of death, on the other
hand, is coded by the International Classifi-
cation of Diseases, which is based primarily
on site of origin rather than histology
especially for solid tumors [3]. For example,
mortality data would use kidney cancer but
the ICCC grouping would be Wilms’ tumor.
Therefore, all of the incidence rates pre-
sented in this chapter are based primarily
on site rather than histology. Note, that
this does not effect non-solid tumors such
as leukemia which would have comparable
groups in each. More incidence and mortal-
ity rates using comparable categories can
be found in the SEER Cancer Statistics
Review: 1973-1996 [6].
Summary
Cancer mortality has declined dramatically
for children. In the United States today
few children die from cancer in comparison
to other causes of death. In 1995, for
children younger than 20 years of age, the
major causes of death were:
• conditions from the perinatal
period (13,449);
• accidents (13,234);
• congenital anomalies (7,949);
• homicides (4,617);
• SIDS (3,397);
• cancer (2,275);
• suicides (2,227 deaths).
Of the nearly 60,000 deaths among
children younger than 20 years of age, less
than 4% were due to neoplasms (cancer). If
infants are excluded, the number one cause
of death was accidents followed by
homicides, suicides and then cancer.
Reference List
1. World Health Organization, International
Classification of Diseases for Oncology, First
Edition, Geneva, 1976.
2. Percy C, Van Holten V, and Muir C, Eds.
International Classification of Diseases for
Oncology, Second Ed., World Health Organiza-
tion, Geneva, 1990.
3. World Health Organization, International
Classification of Diseases, 1975 Revision, vols.1
and 2, Geneva, 1977.
4. Smith MA, et al: Trends in reported incidence
of primary malignant brain tumors in children
in the United States. J Natl Cancer Inst.
90:1269-77, 1998.
5. Kramarova E, Stiller CA: The international
classification of childhood cancer. Int J Cancer:
68:759-65, 1996.
6. Ries LAG, Kosary CL, Hankey BF, Miller BA,
Clegg L, Edwards BK (eds). SEER Cancer
Statistics Review 1973-1996, National Cancer
Institute, http://www-seer.ims.nci.nih.gov,
1998.
Figure XIV.9: Trends in age-specific cancer mortality
rates by type, age 15-19, all races
both sexes, United States, 1975-95
*
*
*
*
*
*
*
#
#
#
#
#
#
#
(
(
(
(
(
(
(
,
,
,
,
,
,
,
&
&
&
&
&
&&
1975-77 1981-83 1987-89 1993-95
Year of death
0
5
10
15
20
25
Average annual rate per million
Brain & ONS
Leukemia
Bones & joints
Soft tissue
Non-Hodgkins lymphomas
&
,
(
#
*

170
National Cancer Institute
SEER Pediatric Monograph

XV
171
National Cancer Institute SEER Pediatric Monograph
OTHER RESOURCES AVAILABLE AT NCI/NIH
Cancer Information Service (CIS) – 1-800-4-CANCER or
http://cis.nci.nih.gov/contact/faqform.html
♦ The Cancer Information Service is the National Cancer Institute’s link to the public,
providing current scientific information in understandable language to patients, their
families, the general public and health professionals. Through a network of 19 regional
offices located throughout the country, the CIS serves the entire United States and
Puerto Rico. CIS staff are available Monday through Friday from 9:00 a.m. to 4:30 p.m.
local time. Callers with TTY equipment may call 1–800–332–8615. Recorded informa-
tion on cancer topics is available 24 hours a day.
NCI publications on childhood cancer: Available by calling 1-800-4-CANCER (1-800-
422-6237).
Also, these publications may be viewed on the Cancer Net’s Kids Home page at
http://cancernet.nci.nih.gov/occdocs/KidsHome.html
♦ Managing Your Child’s Eating Problems: During Cancer Treatment This booklet in-
cludes information about the importance of nutrition, the side effects of cancer and its
treatment, ways to encourage your child to eat, and special diets.
♦ Talking With Your Child About Cancer: This booklet is for a parent whose child has
been diagnosed with cancer. It addresses the health-related concerns of young people of
different ages; it suggests ways to discuss disease-related issues with a child.
♦ When Someone In Your Family Has Cancer: This booklet is for young people whose
parent or sibling has cancer. It includes sections on the disease, its treatment, and
emotional concerns.
♦ Young People With Cancer: A Handbook For Parents: This booklet discusses the most
common types of childhood cancer, treatments, side effects, and issues that may arise
when a child is diagnosed with cancer. It offers medical information and practical tips
gathered from parents.
National Cancer Institute Information (http://cis.nci.nih.gov/resources/resources.html)
♦ The National Cancer Institute is the Federal Government’s principal agency for cancer
research and training. The Web site contains information about the latest news on
cancer research and treatment, upcoming events at NCI, job opportunities, and links to
cancer information resources. A publication index, containing some full-text NCI publi-
cations, is also available.
PDQ
®
(http://cancernet.nci.nih.gov/pdq.htm):
♦ NCI’s comprehensive cancer database, including summaries on cancer treatment,
screening, prevention, and supportive care, and information on ongoing clinical trials.
♦ PDQ
®
is a dynamic database that is updated regularly to ensure that the information it
contains is consistent with the results of the latest cancer research. PDQ
®
contains:

XV
172
National Cancer Institute
SEER Pediatric Monograph
OTHER RESOURCES AVAILABLE AT NCI/NIH
cancer information summaries describing the latest advances in cancer treatment,
supportive care, screening, and prevention; an extensive register of over 1,500 ongoing
clinical trials, with information about studies around the world; and directories of over
23,000 physicians and over 11,000 organizations active in cancer treatment and care.
Most cancer information summaries appear in two versions: a technical version for the
health professional and a non-technical version for patients, their families, and the
public. Both are available in English and Spanish. The information in the database is
peer reviewed by editorial boards of oncology experts and updated monthly.
cancerTrials™ Web site (http://cancertrials.nci.nih.gov/):
♦ NCI’s comprehensive clinical trials information center for patients, health professionals,
and the public. Includes information on understanding trials, deciding whether to
participate in trials, finding specific trials, plus research news and other resources.
CancerNet™ (http://cancernet.nci.nih.gov/index.html):
♦ The NCI International Cancer Information Center’s World Wide Web site, CancerNet™,
provides both health professionals and the public access to a variety of information on
cancer.
MEDLINEplus (http://www.nlm.nih.gov/medlineplus/ )
♦ The National Library of Medicine’s MEDLINEplus Web site includes links to informa-
tion about a number of health topics, medical dictionaries, databases (including
MEDLINE), clearing houses, directories, organizations, publications and health news,
and consumer health libraries.
NCI Office of Liaison Activities:
♦ Throughout the Nation, hundreds of cancer advocacy and outreach organizations pro-
vide education and support to their communities. The Office of Liaison Activities is
NCI’s central point of contact to the national advocacy organizations and, through them,
to the community-based groups. This office maintains ongoing communications and
information exchange between the cancer advocacy organizations and NCI, encouraging
input and feedback from them, and cooperates and collaborates with these groups in
areas of mutual interest. The Office of Liaison Activities serves as a catalyst and re-
source to link advocates with NCI programs, working groups and advisory committees
,
and helps to integrate consumer advocate representatives throughout the NCI. The
Office of Liaison Activities also builds relationships with professional societies and
federal agencies, and provides input and perspective to NCI on complex issues relevant
to cancer patients and the public. Telephone number 301-594-3194 and FAX 301-480-
7558.

XV
173
National Cancer Institute SEER Pediatric Monograph
OTHER RESOURCES AVAILABLE AT NCI/NIH
On the SEER Web page, http://www-seer.ims.nci.nih.gov/, the Cancer Statistics Branch
(CSB) provides additional information on cancer statistics. This monograph can be viewed
at this Web address under Publications. Other SEER publications/monographs (SEER
Cancer Statistics Review (CSR); SEER Prostate Cancer Trends, 1973-1995; Racial/Ethnic
Patterns of Cancer in the United States; and Cancer Rates and Risks) are also included in
this area of the Web page. The CSR is an annual compendium of the most recent cancer
statistcis available. The current version, 1973-1996, includes two chapters on childhood
cancer: one by primary cancer site and a second by the International Classification of
Childhood Cancer.
The CSB provides additional SEER data on the SEER Web page under Scientific Systems:
• CANQUES is an interactive system on the Web that allows the user to access over 10
million pre-calculated cancer statistics.
• A SEER public-use file with SEER*Stat provides an easy to use PC desktop system for
the production of a myriad of cancer statistics such as incidence rates, survival rates by
various demographic and tumor variables. This CD-ROM can be ordered from the Web
page.
The Applied Research Branch (ARB) Web site,
http://www-dccps.ims.nci.nih.gov/ARB/index.html, provides information on the following
topics: Risk Factor Monitoring & Methods, SEER-Medicare, Cancer Statistics Methods &
Models, Dietary Assessment, Breast Cancer Surveillance Consortium, Health Services &
Economics, HMO Cancer Research Network, Outcomes Research. Under Cancer Statistics
Methods & Models, there is information about the methodology and estimation for cancer
prevalence and the probability of developing cancer.

174
National Cancer Institute
SEER Pediatric Monograph

175
National Cancer Institute SEER Pediatric Monograph
INTERNATIONAL CLASSIFICATION OF CHILDHOOD CANCER (ICCC)
Source: Kramárová E, Stiller CA, Ferlay J, Parkin DM, Draper GJ, Michaelis J,
Neglia J, Qureshi S (1996) International Classification of Childhood
Cancer1996. IARC Technical Report No.29, International Agency for
Research of Cancer, Lyon.
ICCC GROUP MORPHOLOGY TOPOGRAPHY
I Leukemia
(a) Lymphoid Leukemia
Excluding ALL 9820, 9822-9827, 9850 C00.0-C80.9
ALL 9821 C00.0-C80.9
(b) Acute Leukemia
Excluding AML 9840, 9841, 9864, 9866, 9867, 9891,
9894, 9910
C00.0-C80.9
AML 9861 C00.0-C80.9
(c) Chronic Myeloid
Leukemia
9863, 9868 C00.0-C80.9
(d) Other Specified
Leukemias
9830, 9842, 9860, 9862, 9870-9890,
9892, 9893, 9900, 9930-9941
C00.0-C80.9
(e) Unspecified Leukemias 9800-9804 C00.0-C80.9
II Lymphomas and
Reticuloendothelial
Neoplasms
(a) Hodgkin’s disease 9650-9667 C00.0-C80.9
(b) Non-Hodgkin’s
lymphomas
9591-9595, 9670-9686, 9690-9717,
9723, 9688
C00.0-C80.9
(c) Burkitt’s lymphoma 9687 C00.0-C80.9
(d) Miscellaneous
lymphoreticular
neoplasms
9720, 9731-9764 C00.0-C80.9
(e) Unspecified lymphomas 9590 C00.0-C80.9
III CNS and
Miscellaneous
Intracranial and
Intraspinal Neoplasms
(a) Ependymoma 9383, 9390-9394 C00.0-C80.9
(b) Astrocytoma 9380 C72.3
9381, 9400-9441 C00.0-C80.9
(c) Primitive
neuroectodermal tumors
9470-9473 C00.0-C80.9*
(d) Other gliomas 9380 C70.0-C72.2, C72.4-
C72.9
9382, 9384, 9442-9460, 9481 C00.0-C80.9
(e) Miscellaneous
intracranial and
intraspinal neoplasms
8270-8281, 8300, 9350-9362, 9480,
9505, 9530-9539
C00.0-C80.9
(f) Unspecified intracranial
and intraspinal
neoplasms
8000-8004 C70.0-C72.9, C75.1-
C75.3
*For this monograph, any cases with site codes C00.0-C69.9, C73.9-C75.0, C75.4-C77.9
were removed from this group.

INTERNATIONAL CLASSIFICATION OF CHILDHOOD CANCER (ICCC)
176
National Cancer Institute
SEER Pediatric Monograph
Source: Kramárová E, Stiller CA, Ferlay J, Parkin DM, Draper GJ, Michaelis J,
Neglia J, Qureshi S (1996) International Classification of Childhood
Cancer1996. IARC Technical Report No.29, International Agency for
Research of Cancer, Lyon.
ICCC GROUP MORPHOLOGY TOPOGRAPHY
IV Sympathetic Nervous
System Tumors
(a) Neuroblastoma and
ganglioneuroblastoma
9490, 9500 C00.0-C80.9
(b) Other sympathetic
nervous system tumors
8680, 8693-8710, 9501-9504, 9520-
9523
C00.0-C80.9
V Retinoblastoma
9510-9512 C00.0-C80.9
VI Renal Tumors
(a) Wilms’ tumor, rhabdoid
and clear cell sarcoma
8963 C64.9, C80.9
8960, 8964 C00.0-C80.9
(b) Renal carcinoma 8010-8041, 8050-8075, 8082, 8120-
8122, 8130-8141, 8143, 8155, 8190-
8201, 8210, 8211, 8221-8231,8240,
8241, 8244-8246, 8260-8263, 8290,
8310, 8320, 8323, 8401, 8430, 8440,
8480-8490, 8504, 8510, 8550, 8560-
8573
C64.9
8312 C00.0-C80.9
(c) Unspecified malignant
renal tumors
8000-8004 C64.9
VII Hepatic Tumors
(a) Hepatoblastoma 8970 C00.0-C80.9
(b) Hepatic carcinoma 8010-8041, 8050-8075, 8082, 8120-
8122, 8140, 8141, 8143, 8155, 8190-
8201, 8210, 8211, 8230, 8231,8240,
8241, 8244-8246, 8260-8263, 8310,
8320, 8323, 8401, 8430, 8440, 8480-
8490, 8504, 8510, 8550, 8560-8573
C22.0, C22.1
8160-8180 C00.0-C80.9
(c) Unspecified malignant
hepatic tumors
8000-8004 C22.0, C22.1
VIII Malignant Bone
Tumors
(a) Osteosarcoma 9180-9200 C00.0-C80.9
(b) Chrondosarcoma 9220-9230 C00.0-C80.9
9231, 9240 C40.0-C41.9
(c) Ewing’s sarcoma 9260 C40.0-C41.9, C80.9
9363, 9364 C40.0-C41.9
(d) Other specified
malignant bone tumors
8812, 9250, 9261-9330, 9370 C00.0-C80.9
(e) Unspecified malignant
bone tumors
8000-8004, 8800, 8801, 8803, 8804 C40.0-C41.9

177
National Cancer Institute SEER Pediatric Monograph
INTERNATIONAL CLASSIFICATION OF CHILDHOOD CANCER (ICCC)
Source: Kramárová E, Stiller CA, Ferlay J, Parkin DM, Draper GJ, Michaelis J,
Neglia J, Qureshi S (1996) International Classification of Childhood
Cancer1996. IARC Technical Report No.29, International Agency for
Research of Cancer, Lyon.
ICCC GROUP MORPHOLOGY TOPOGRAPHY
IX Soft-Tissue Sarcomas
(a) Rhabdomyosarcoma and
embryonal sarcoma
8900-8920, 8991 C00.0-C80.9
(b) Fibrosarcoma,
neurofibrosarcoma and
other
fibromatous neoplasms
8810, 8811, 8813-8833, 9540-9561 C00.0-C80.9
(c) Kaposi’s sarcoma 9140 C00.0-C80.9
(d) Other specified soft-
tissue sarcomas
8840-8896, 8982, 8990, 9040-9044,
9120-9134, 9150-9170, 9251, 9581
C00.0-C80.9
8963 C00.0-C63.9, C65.9-
C76.8
9231, 9240, 9363, 9364 C00.0-C39.9, C44.0-
C80.9
9260 C00.0-C39.9, C47.0-
C76.8
(e) Unspecified soft-tissue
sarcomas
8800-8804 C00.0-C39.9, C44.0-
C80.9
X Germ-Cell,
Trophoblastic and other
Gonadal Neoplasms
(a) Intracranial and
intraspinal germ-cell
tumors
9060-9102 C70.0-C72.9, C75.1-
C75.3
(b) Other and unspecified
non-gonadal germ-cell
tumors
9060-9102 C00.0-C55.9, C57.0-
C61.9, C63.0-C69.9,
C73.9-C75.0, C75.4-
C80.9
(c) Gonadal germ-cell
tumors
9060-9102 C56.9, C62.0-C62.9
(d) Gonadal carcinomas 8010-8041, 8050-8075, 8082, 8120-
8122, 8130-8141, 8143, 8155, 8190-
8201, 8210, 8211, 8221-8241, 8244-
8246, 8260-8263, 8290, 8310, 8320,
8323, 8430, 8440, 8480-8490, 8504,
8510, 8550, 8560-8573
C56.9, C62.0-C62.9
8380, 8381, 8441-8473 C00.0-C80.9
(e) Other and unspecified
malignant gonadal
tumors
8590-8670, 9000 C00.0-C80.9
8000-8004 C56.9, C62.0-C62.9
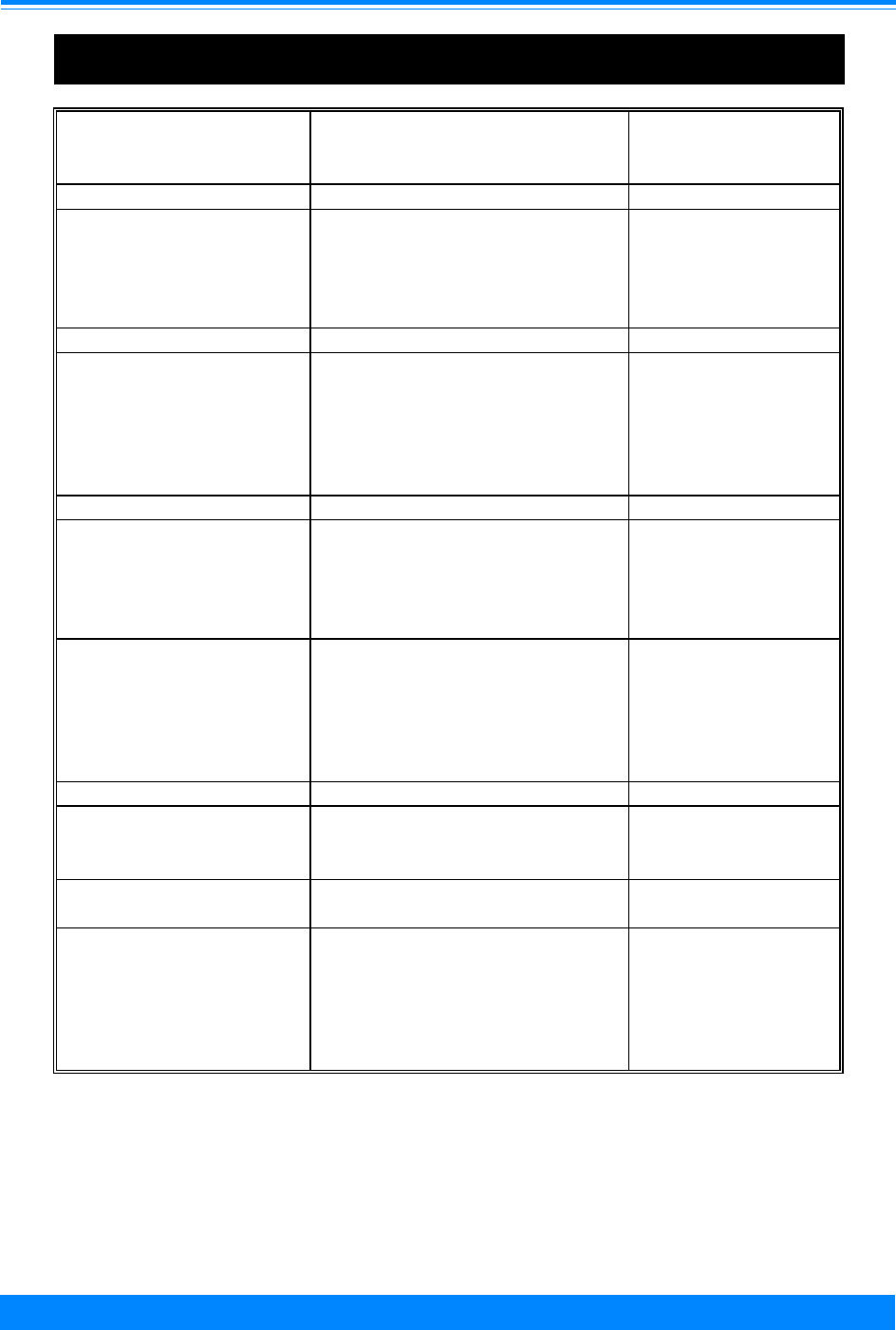
INTERNATIONAL CLASSIFICATION OF CHILDHOOD CANCER (ICCC)
178
National Cancer Institute
SEER Pediatric Monograph
Source: Kramárová E, Stiller CA, Ferlay J, Parkin DM, Draper GJ, Michaelis J,
Neglia J, Qureshi S (1996) International Classification of Childhood
Cancer1996. IARC Technical Report No.29, International Agency for
Research of Cancer, Lyon.
ICCC GROUP MORPHOLOGY TOPOGRAPHY
XI Carcinomas and other
Malignant Epithelial
Neoplasms
(a) Adrenocortical carcinoma 8370-8375 C00.0-C80.9
(b) Thyroid carcinoma 8010- 8041, 8050-8075, 8082, 8120-
8122, 8130-8141, 8155, 8190, 8200,
8201, 8211, 8230, 8231, 8244-8246,
8260-8263, 8290, 8310, 8320, 8323,
8430, 8440, 8480, 8481, 8500-8573
C73.9
8330-8350 C00.0-C80.9
(c) Nasopharyngeal
carcinoma
8010- 8041, 8050-8075, 8082, 8120-
8122, 8130-8141, 8155, 8190, 8200,
8201, 8211, 8230, 8231, 8244-8246,
8260-8263, 8290, 8310, 8320, 8323,
8430, 8440, 8480, 8481, 8504, 8510,
8550, 8560-8573
C11.0-C11.9
(d) Malignant melanoma 8720-8780 C00.0-C80.9
(e) Skin carcinoma 8010- 8041, 8050-8075, 8082, 8090-
8110, 8140, 8143, 8147, 8190, 8200,
8240, 8246, 8247, 8260, 8310, 8320,
8323, 8390-8420, 8430, 8480, 8542,
8560, 8570-8573, 8940
C44.0-C44.9
(f) Other and unspecified
carcinomas
8010-8082, 8120-8155, 8190-8263,
8290, 8310, 8314-8323, 8430-8440,
8480-8580, 8940, 8941
C00.0-C10.9, C12.9-
C21.8, C23.9-C39.9,
C48.0-C48.8, C50.0-
C55.9, C57.0-C61.9,
C63.0-C63.9, C65.9-
C72.9, C75.0-C80.9
XII Other and
Unspecified Malignant
Neoplasms
(a) Other specified
malignant tumors
8930, 8933, 8950, 8951, 8971-8981,
9020, 9050-9053, 9110, 9580
C00.0-C80.9
(b) Other unspecified
malignant tumors
8000-8004 C00.0-C21.8, C23.9-
C39.9, C42.0-C55.9,
C57.0-C61.9, C63.0-
C63.9, C65.9-C69.9,
C73.9-C75.0, C75.4-
C80.9

179
National Cancer Institute SEER Pediatric Monograph
INDEX
Topic Chapter
Acute erythremia I - Table I.1
Acute lymphoblastic leukemia (ALL) I
Acute myelofibrosis I - Table I.1
Acute myeloid leukemia (AML) I
Acute non-lymphocytic leukemia I
Acute panmyelosis I - Table I.1
Adrenal gland IV
Adrenocortical carcinoma XI
Age-adjusted rate (definition) Intro
Age-specific rate (definition) Intro
ALL (Acute lymphoblastic leukemia) I
Alveolar rhabdomyosarcoma IX
AML (Acute myeloid leukemia) I
Angiosarcoma IX
Astrocytomas III
Astrocytomas in infants Infants
Bilateral retinoblastoma V
Bone VIII
Bone tumors in infants Infants
Burkitt's lymphoma II
Cancer, liver VII
Cancer, renal VI
Cancers, renal VI
Case-control study (definition Intro
Central nervous system (CNS) III
Central nervous system malignancies III
Chondrosarcoma IX
Chondrosarcoma VIII
Chronic erythremia I - Table I.1
Chronic myeloid leukemia (CML) I
CML (Chronic myeloid leukemia) I
CNS (Central nervous system) III
CNS malignancies in infants Infants
CNS tumors among adolescents 15-19 year olds
Cohort study (definition) Intro
Dermatofibrosarcoma IX
Dysgerminomas X
EAPC (Estimated annual percent change) (definition) Intro
Embryonal rhabdomyosarcoma IX
Ependymoma III
Erythremia, acute I - Table I.1
Erythremia, chronic I - Table I.1
Estimated annual percent change (EAPC) (definition) Intro
Etiology, Multifactorial (definition) Intro
Ewing's (extraosseous) sarcoma IX
Ewing's sarcoma VIII

180
National Cancer Institute
SEER Pediatric Monograph
INDEX
Topic Chapter
Eye, retina V
Fibrosarcoma IX
Germ cell tumor X
Germ cell tumor, gonadal X
Germ cell tumor, intracranial X
Germ cell tumor, Intraspinal X
Germ cell tumor, non-gonadal X
Germ cell tumor, ovarian X
Germ cell tumor, testicular X
Germ cell tumors among adolescents 15-19 year olds
Germ cell tumors in infants Infants
Gliomas III
Gonadal carcinoma X
Gonadal germ cell tumor X
Gonadal tumor X
HD (Hodgkin's Disease) II
Hemangiopericytoma, malignant IX
Hemangiosarcoma IX
Hepatic carcinomas VII
Hepatic tumors VII
Hepatic tumors in infants Infants
Hepatoblastoma VII
Hepatocellular carcinoma VII
Histiocytoma, malignant fibrous IX
Histologic confirmation Intro
Hodgkin's Disease (HD) II
Hodgkin's disease among adolescents 15-19 year olds
ICCC Classification (International Classification of Childhood Cancers) Intro
Incidence (definition) Intro
Infants Infants
International Classification of Childhood Cancers (ICCC) Intro
Intracranial germ cell tumor X
Intraspinal germ cell tumor X
Juvenile chronic myelomonocytic leukemia (JMML) Infants
Kaposi's sarcoma IX
Kindey VI
Leiomyosarcoma IX
Leukemia I
Leukemia in infants Infants
Leukemias I
Liposarcoma IX
Liver cancer VII
Lymphoctytic depletion II
Lymphocytic predominance II
Lymphoid leukemia I
Lymphoma II
Lymphomas II
Lymphomas in infants Infants
Lymphoreticular neoplasms II
Melanoma XI

181
National Cancer Institute SEER Pediatric Monograph
INDEX
Topic Chapter
Melanoma among adolescents 15-19 year olds
Mixed cellularity II
Mortality Mortality
Mortality rate (definition) Intro
Myelofibrosis, acute I - Table I.1
Myeloid sarcoma I - Table I.1
Nasopharyngeal carcinoma XI
Neoplasms, lymphoreticular II
Nerve sheath tumor, malignant peripheral IX
Neuroblastoma in infants Infants
Neuroblastomas IV
NHL (non-Hodgkin's lymphoma) II
Nodular scerosis II
non-Hodgkin's lymphoma (NHL) II
Non-Hodgkin's lymphoma among adolescents 15-19 year olds
Osteosarcoma VIII
Ovarian germ cell tumor X
Ovary X
Panmyelosis, acute I - Table I.1
PNET (Primitive neuroectodermal tumors) III
Population Intro
Primitive neuroectodermal tumors (PNET) III
Reed-Sternberg II
Relative risk (definition) Intro
Relative survival (definition) Intro
Renal cancers VI
Retina of the eye V
Retinoblastoma in infants Infants
Retinoblastomas V
Rhabdomyosarcoma (RMS) IX
Risk factor (definition) Intro
RMS (Rhabdomyosarcoma) IX
Rye classification II
Sarcoma, myeloid I - Table I.1
Sarcoma, soft tissue IX
Sarcomas of the bone VIII
Sarcomas, soft tissue IX
SEER (Surveillance, Epidemiology and End Results) Intro
Skin carcinoma XI
Soft tissue sarcomas IX
Soft tissue sarcomas in infants Infants
Surveillance, Epidemiology and End Results (SEER) Intro
Sympathetic nervous system IV
Synovial sarcoma IX
Teratomas X
Testicular germ cell tumor X
Testis X
Thyroid cancer XI
Thyroid cancer among adolescents 15-19 year olds
Thyroid carcinomas XI

182
National Cancer Institute
SEER Pediatric Monograph
INDEX
Topic Chapter
Trophoblastic tumor X
Wilms' tumor VI
Wilms' tumor in infants Infants
Yolk sac tumors X
